
This post comes out a recent conversation I had with Tucker Goodrich and David Gornoski (not live yet, I’ll post it when its out). Tucker was arguing that obesity was caused by endogenous cannabinoids that are made from linoleic acid. I was arguing that obesity is caused by reductive stress. As I pointed out on the show, these are not mutually exclusive ideas. They are in fact self-reinforcing. Reductive stress drives endocannabinoid production.
Anandamide
Anandamide (AEA) is an endogenous cannabinoid (it triggers a cannabis receptor). In lean humans it rises before a meal and then falls after the meal is taken.1 Obese humans have higher fasting levels of AEA and a lesser drop in AEA after a meal.
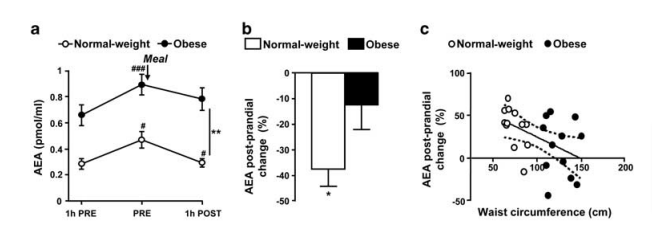
Of course cannabis (weed, marijuana, pot, reefer) is known for giving people the munchies. Elevated AEA is likely to increase one’s appetite, especially if it remains elevated after a meal.
Furthermore, mice who lack the cannabinoid receptor CB1 – the one that is activated by AEA – eat less and are resistant to obesity.
EAE is made from linoleic acid
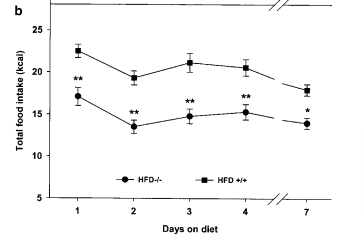
EAE is made from linoleic acid PUFA. Linoleic acid is converted to arachidonic acid, which is then converted to AEA. If eat a lot of linoleic acid, it can increase AEA.
Mice who are fed 8% of calories as linoleic acid have more AEA than mice fed 1% and they get fat.2 We’ll come back to the ones that were fed fish oil (EPA and DHA).
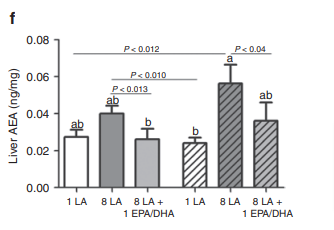
D6D and D5D control Arachidonic Acid production
Before linoleic acid can be made into EAE, it has to first be made into Arachindonic acid, which has two more double bonds than linoleic acid. The rate-limiting enzymes that control the conversion from linoleic to arachidonic acid are called delta 6 desaturase (D6D) and delta 5 desaturase (D5D). Arachidonic acid can then be used to make AEA.
D6D adds a double bond to linoleic acid, transforming it to gamma linolenic acid. If we look at the ratio of linoleic acid to gamma linolenic acid, it gives us an indirect indication of the activity level of the enzyme D6D.
The ratio of linoleic acid to gamma linolenic acid is called the D6D desaturase ratio and it is associated with obesity in actual, walking around humans.3 If you make a lot of D6D, you will convert a lot of linoleic acid to not only AEA, but also oxidized linoleic acid metabolites such as 12-HETE4, etc.
Having high D6D activity seems quite bad. High D6D activity is a VERY strong predictor of diabetes risk.5 In the Potsdam study, in the group with the lowest D6D activity 47 people developed diabetes. In the group with the highest D6D activity, 264 people developed diabetes. Additionally, having a more active genetic variant of the D6D enzyme is causally linked to diabetes risk.6
NADH Controls D6D (and D5D and SCD1) Activity
As I’ve already mentioned, your genetics and dietary linoleic acid affect D6D activity. But you can’t change your genes. And if you are a longtime reader of this blog, I hope you’re already avoiding linoleic acid. But it’s impossible to avoid it entirely. Even Firebrand Meats low-PUFA pork still has 6% linoleic acid.
A recent paper7 showed the third major thing that drives D6D and D5D (and presumably SCD1) activity: high cytoplasmic NADH levels (reductive stress). To help understand this, let’s look at how all desaturase reactions work.
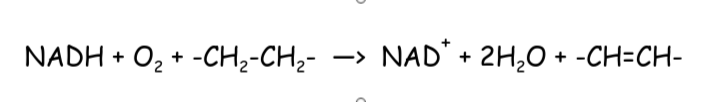
This is the reaction for all three desaturases – SCD1, D6D and D5D. The -CH2-CH2- represents a saturated bond and -CH=CH- represents an unsaturated bond. NADH is a cofactor of desaturase reactions. When levels of cofactors and substrates (the thing being acted on by the enzyme, in D6D’s case linoleic acid) are high, the reaction proceeds at a faster rate. When linoleic acid and NADH are both high, D6D will work very quickly.
Conversely, enzymes are slowed down by their end products to avoid them running out of control. So SCD1 is slowed down by high NAD+ and it’s end-product palmitoleic acid. D6D and D5D are slowed down by their end products EPA and DHA. When the ratio of NADH/NAD+ is high (reductive stress), the desaturases will work at a fast pace.
Interestingly, if you remove any of the three desaturase enzymes from a mouse, they become resistant to obesity.8–10 Desaturase activity is necessary for obesity. Reductive stress drives desaturase activity.
Obese Humans are in Reductive Stress
Thinking back to the obese humans who produce high levels of AEA both before and after a meal, it is logical to ask if they are in redductive stress. They are.11
This is a graph of lean vs. obese people eating a high carb meal. Lower numbers means a higher NADH/NAD+ ratio (lower is bad). The lean people are the open circles and the obese are the filled circles. The obese group has higher NADH/NAD+ levels at baseline. They are in reductive stress. They will have high D6D activity and make a lot of AEA. When the lean people eat they have a brief spike in NAD+ levels that is blunted in the obese. By around 2 hours after the meal, the NADH/NAD+ ratio rises in both groups (the line descends), but the lean group never gets as high as the obese group at any point.
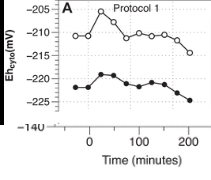
The obese group fails to get out of reductive stress and has consistently elevated AEA.
Why are the obese in reductive stress? It is likely to be a combination of elevated SCD112 activity and elevated PARP as you will soon see in the twin study. This is a positive feedback loop. High NADH/NAD+ levels drive the desaturase enzymes, which push the NADH/NAD+ ratio higher (over time, in the short term they do supply a little NAD+).
High Linoleic Acid Protects from Diabetes
The Potsdam study5 also showed that high levels of linoleic acid and arachidonic acid are both very protective in predicting diabetes risk. Furthermore, low levels of linoleic acid in cholesterol esters are associated with obesity3. How do we explain these paradoxes?
Understanding the role of D6D and D5D helps us understand the paradox. The levels of linoleic acid in the body is only mildly associated with dietary intake5: its levels are mostly controlled by D6D, D5D and downstream enzymes in a population where everyone eats a very similar diet.13 If you have a lot of linoleic acid and arachidonic acid, that means you’re not converting them to AEA and 12-HETE.
If you convert them to AEA, you’ll have the munchies. If you convert them to 12-HETE, you’ll activate the Aryl Hydrocarbon Receptor14, which will upregulate PARP15, an enzyme which lowers levels of NAD+, contributing to the reductive stress and driving the desaturase enzymes. In fact, in a study of identical twins where one was obese and the other was lean, a major finding was upregulated PARP and high NADH/NAD+ levels in the obese twin.16 This wasn’t caused by genetics or epigenetics.
Conclusion
Endogenous cannabinoid production contributes to dysregulated appetite. D6D is the limiting enzyme in the production of cannabinoids and oxylipins, both of which are associated with obesity and diabetes. D6D activity level is driven by a high NADH/NAD+ ratio (reductive stress).
Genetics, dietary linoleic acid (and EPA and DHA) and NADH/NAD+ levels all contribute to D6D activity. You can’t change your genetics, but you can change your diet. Avoid linoleic acid and consider increasing EPA and DHA. Use stearic acid with meals to slow down fat absorption and flow.
Alpha-lipoic acid fights reductive stress directly. Sterculia Oil fights the runaway feedback loop of reductive stress driving SCD1 activity which drives reductive stress.
I’ve got more ideas to fight reductive stress in the works, check back soon.
- 1.Gatta-Cherifi B, Matias I, Vallée M, et al. Simultaneous postprandial deregulation of the orexigenic endocannabinoid anandamide and the anorexigenic peptide YY in obesity. Int J Obes. Published online August 16, 2011:880-885. doi:10.1038/ijo.2011.165
- 2.Alvheim AR, Malde MK, Osei-Hyiaman D, et al. Dietary Linoleic Acid Elevates Endogenous 2-AG and Anandamide and Induces Obesity. Obesity. Published online October 2012:1984-1994. doi:10.1038/oby.2012.38
- 3.Warensjö E, Öhrvall M, Vessby B. Fatty acid composition and estimated desaturase activities are associated with obesity and lifestyle variables in men and women. Nutrition, Metabolism and Cardiovascular Diseases. Published online March 2006:128-136. doi:10.1016/j.numecd.2005.06.001
- 4.Deol P, Fahrmann J, Yang J, et al. Omega-6 and omega-3 oxylipins are implicated in soybean oil-induced obesity in mice. Sci Rep. Published online October 2, 2017. doi:10.1038/s41598-017-12624-9
- 5.Kröger J, Zietemann V, Enzenbach C, et al. Erythrocyte membrane phospholipid fatty acids, desaturase activity, and dietary fatty acids in relation to risk of type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition (EPIC)–Potsdam Study. The American Journal of Clinical Nutrition. Published online October 27, 2010:127-142. doi:10.3945/ajcn.110.005447
- 6.Jäger S, Cuadrat R, Hoffmann P, Wittenbecher C, Schulze MB. Desaturase Activity and the Risk of Type 2 Diabetes and Coronary Artery Disease: A Mendelian Randomization Study. Nutrients. Published online July 28, 2020:2261. doi:10.3390/nu12082261
- 7.Kim W, Deik A, Gonzalez C, et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD+ Recycling. Cell Metabolism. Published online April 2019:856-870.e7. doi:10.1016/j.cmet.2018.12.023
- 8.Powell D, Gay J, Smith M, et al. Fatty acid desaturase 1 knockout mice are lean with improved glycemic control and decreased development of atheromatous plaque. DMSO. Published online June 2016:185. doi:10.2147/dmso.s106653
- 9.Stoffel W, Hammels I, Jenke B, et al. Obesity resistance and deregulation of lipogenesis in Δ6‐fatty acid desaturase ( <scp>FADS</scp> 2) deficiency. EMBO Rep. Published online December 30, 2013:110-120. doi:10.1002/embr.201338041
- 10.Miyazaki M, Sampath H, Liu X, et al. Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochemical and Biophysical Research Communications. Published online March 2009:818-822. doi:10.1016/j.bbrc.2009.01.183
- 11.Istfan N, Hasson B, Apovian C, et al. Acute carbohydrate overfeeding: a redox model of insulin action and its impact on metabolic dysfunction in humans. American Journal of Physiology-Endocrinology and Metabolism. Published online November 1, 2021:E636-E651. doi:10.1152/ajpendo.00094.2021
- 12.Dziewulska A, Dobosz AM, Dobrzyn A, et al. SCD1 regulates the AMPK/SIRT1 pathway and histone acetylation through changes in adenine nucleotide metabolism in skeletal muscle. Journal Cellular Physiology. Published online June 26, 2019:1129-1140. doi:10.1002/jcp.29026
- 13.Chen X, Scholl TO, Leskiw M, Savaille J, Stein TP. Differences in Maternal Circulating Fatty Acid Composition and Dietary Fat Intake in Women With Gestational Diabetes Mellitus or Mild Gestational Hyperglycemia. Diabetes Care. Published online September 1, 2010:2049-2054. doi:10.2337/dc10-0693
- 14.Chiaro ChristopherR, Patel RD, Perdew GaryH. 12(R)-Hydroxy-5(Z),8(Z),10(E),14(Z)-eicosatetraenoic Acid [12(R)-HETE], an Arachidonic Acid Derivative, Is an Activator of the Aryl Hydrocarbon Receptor. Mol Pharmacol. Published online September 8, 2008:1649-1656. doi:10.1124/mol.108.049379
- 15.Diani-Moore S, Shoots J, Singh R, Zuk JB, Rifkind AB. NAD+ loss, a new player in AhR biology: prevention of thymus atrophy and hepatosteatosis by NAD+ repletion. Sci Rep. Published online May 23, 2017. doi:10.1038/s41598-017-02332-9
- 16.Jukarainen S, Heinonen S, Rämö JT, et al. Obesity Is Associated With Low NAD+/SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. The Journal of Clinical Endocrinology & Metabolism. Published online January 2016:275-283. doi:10.1210/jc.2015-3095
Hi Brad,
I have to admit I am beginning to become confused on a few bits as it does appear you have had a shift of thought process. I have listed what my own current understanding on things is and have added a couple of questions at the bottom. I would be eternally grateful if you could please help me to clear things up again:
Carbs:
My current understanding / tactics with carbs are as follows:
I keep starch in my diet and consume it with both my lunch / dinner (avoiding it at breakfast to avoid the high insulin / high cortisol issue as cortisol is highest in the morning). My understanding was that even if starch is stored as fat that the fat stored is extremely saturated so fine and also that SCD1 isn’t increased a lot in comparison to sugar. I currently consume about 100g – 125g of carbs with at least 80% of that being starch. Sugar I tend to avoid as much as possible mainly because I fear it raises SCD1.
Fat:
I tend to eat strictly saturated fat as much as I can enforce. I have something around the lines of 90g – 120g a day mainly coming from my lunch and dinner as I like to keep my breakfasts lowish fat and low carb with a large amount of protein. My thoughts were also that I am creating 2 big fat blasts with my lunch and dinner which also included a glass of succinade before each of them.
Questions:
You seem to be leaning towards the Ray Peat way of thinking these days and to me he seems to promote higher sugar, higher protein with low to moderate starch and low to moderate fat focused on saturated fats. Would you now say this is how your own thinking is beginning to turn??
Am I wrong not to include carbs with my breakfast?? (is the high insulin / high cortisol issue not as dangerous as I think it is?)
Is combining high fat with high carbs good or bad?? I always thought of it as creating an energy overload to generate a mass of heat but the Ray Peat thing makes me think overwise. Where do you stand on this at the moment?? I think I saw that you have recently said about high post prandial lipolysis maybe not being as great as first thought.
Thanks again for all of your hard work Brad, you are helping a lot of people and you should be proud of yourself!!
As to the starch vs. sugar question and Ray Peaty thinking:
I still think starch is the preferred carbohydrate. Sugar, as you said, raises SCD1 and it makes lab animals fat. I agree with Peat that reductive stress is at the root of the problem but I disagree with sugar as a cure.
I don’t necessarily think that combining fat with starch is bad, I just think that if you DO combine them, it’s best with a high stearic acid fat. The British chef Elizabeth David wrote in French Provincial Cooking, “For deep frying, the French prefer the dripping from beef kidney fat (suet) to any other”. Beef suet is between 23 and 40% stearic acid, depending on the study. This is very high.
As to a high protein breakfast… You will have increased cortisol, but since there is no carbohydrate in it, it’s not clear to me that there’s much harm since you should not have much blood glucose.
Thank you!
Brad
I’ll say as one of the resident starch-eater conversions, sugar still makes me fat & gives me that “fat & dumb happy” (anandamide?) feeling.
I often wonder what the deal is with potheads.
I read a lot about how cannabanoids can lead to overeating and/or obesity. And, a lot of the stuff that you post about linoleic acid and obesity (at least the stuff that doesn’t go over my head) makes sense.
BUT, i know a ton of potheads, who live on high Omega-6 junk food, and are baked 24/7. For every one of them who is overweight, i know 5 who are lean or only carrying around an extra 5 or 10 pounds.
My brother and I are a good example. He is a pothead, i never touch the stuff.
He drinks 1,000 calories a day (minimum) of sugary coffee with half n half, he drinks whole milk instead of water and whatever he eats for lunch and dinner is usually fast food, but when it’s home cooked, it’s cooked in seed oil or margarine. He is only not stoned, when the weed wears off when he’s asleep.
He’s been thin his entire life. He turns 41 in November and he is just now starting to get that 5 to 10 extra pounds Dad bod.
I’m eating a high stearic acid carnivore diet, i drink my coffee black, i put heavy cream (no sweetener) in my pu’erh tea, i take ALA, I’m 42, I’m 6′ 1″ and i can’t seem to get below 240.
Seems so counterintuitive
There are many cannabinoids and they probably have different/opposing effects. I’m focusing on AEA because of it’s clear role in signalling hunger, but that doesn’t tell the whole story of cannabis.
Brad
Adding to the discussion some talk about thyroid function with CB1 receptor at least-
https://pubmed.ncbi.nlm.nih.gov/18755884/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2630898/
One should ask whether CB1 is being downregulated in some potheads (due to CBD or other cannabinoids coming in alongside THC?), as this seems to be right in the direct line of the thyroid regulatory circuit. Trigger CB1 a lot, TSH is downregulated; inhibit CB1 receptors and TSH is upregulated if I’m reading those right.
Your brother is likely “skinny fat” aka TOFI (thin outside, fat inside): https://en.wikipedia.org/wiki/TOFI
I will venture a guess that basic lab tests would confirm significant insulin resistance if not outright prediabetes or diabetes (fasting insulin, A1c, c-peptide, lipid panel, comprehensive metabolic panel, etc).
For your own case, I recommend trying the PE diet approach from Dr. Ted Naiman.
http://www.PtoER.com
http://www.burnfatnotsugar.com
Full book: https://b-ok.cc/book/5281356/6e441e support the authors if you can!
Would backloading carbs be useful (and going heavy on stearic acid in the morning and afternoon) be a useful strategy for staying out of reductive stress? Reason being: insulin and cortisol are highest in the morning, so keeping those low makes sense. Stearic acid creates the buffer to allow fats to not enter too quickly. Carbs are for hormone optimization.
Second point: This makes so much sense. You’re hungry because you have low energy. Snack foods all have linoleic acid, which CAN convert to AA, or enter the mitochondria and cause reductive stress. Mobilizing LA for fuel steals nad+ and creates the NADH buildup more rapidly than stearic acid, which in turn synthesizes AA to stimulate more hunger through the AEA pathway. Basically eat Linoleic Acid and eventually wind up in a state of constant hunger because the foods you’re eating are constantly stealing energy, which requires more “energy” to get you of out of the energy deficit.
So D6D is LA/GLA. or GLA/LA?
GLA/LA, which gives an annoyingly small number like 0.006.
That makes sense
I think you have a typo with the word “protective” in this sentence and meant to use “effective”:
The Potsdam study5 also showed that high levels of linoleic acid and arachidonic acid are both very protective in predicting diabetes risk
Actually, I do mean protective here. In the sense that if you have high levels of linoleic and arachidonic, you are unlikely to get diabetes. This is paradoxical because high levels of LA and AA mean low levels of OXLAMs in a population eating the standard diet.