
My last posts were viewed by some as pessimistic in a “once you’re obese, you are stuck” kind of way. I don’t believe that and very soon I will start writing about solutions. Having said that, this post will perhaps seem pessimistic if you thought the previous ones were. I’m working through the basic mechanisms that control the switch into obesity and a torpid metabolism. Once we have understanding, then we can think about solutions.
This post will complete the chain of events that begins with consuming polyunsaturated fats (PUFA) and ends in obesity and torpor. It also connects the dots between obesity and organic pollutants.
I have said that PPAR gamma is the Gatekeeper that controls entry into torpor and that oxidized PUFA is the key that unlocks it. What I haven’t talked about is how the PUFA gets oxidized. The thing about PUFA is that it can and does oxidize on its own but in the human body it is MUCH more likely to be oxidized by an enzyme.
The enzymes COX1 and COX2 convert arachidonic acid – derived from linoleic acid -into prostanoids. Prostanoids are oxidized PUFA which play a role in inflammation. COX1 and COX2 are blocked by aspirin and ibuprofen. If you eliminate the oxidized PUFA, your headache goes away.
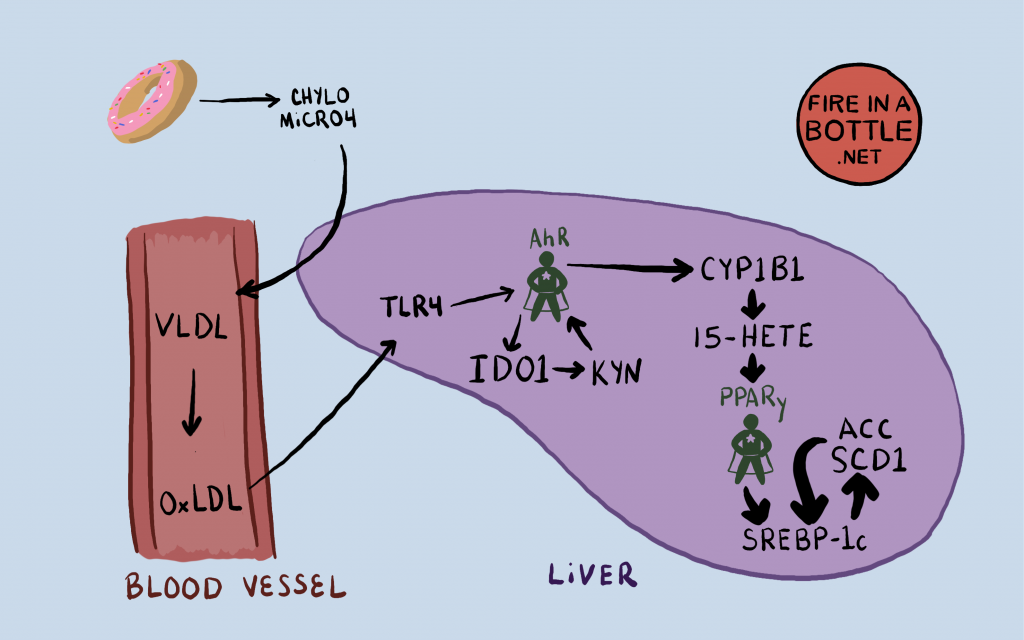
The Aryl Hydrocarbon Receptor (AhR)
This unfortunately named “nuclear receptor” isn’t even really a receptor in the classical sense. It’s not on the cell surface, it lives in the cytosol (inside the cell) and when it binds it’s “ligand” – the thing it recognizes and which turns it on – it goes into the nucleus and turns on other genes.
One of its functions is to help break down environmental toxins. It does this by turning on several genes which code for oxidizing enzymes such as the cytochrome P450s, including the genes CYP1A1, CYP1A2 and CYP1B1.1 Cytochrome P450s oxidize things. We break down toxins by oxidizing them.
For instance, the AhR is turned on by the polycyclic aryl hydrocarbons found in roasted coffee and CYP1A2 is responsible for caffeine metabolism. Polymorphisms in the CYP1A2 region are associated with increased coffee consumption. I am a coffee addict, as is my whole family, which means we likely have an active form of CYP1A2.2
CYP1B1 Turns Arachidonic acid into known PPAR gamma activators
This study3 showed that in the laboratory, CYP1B1 in the presence of NADPH turns arachidonic acid – a downstream product of linoleic acid – into known PPAR gamma ligands (activators) 15-HETE4 and 12-HETE5, among others. CYP1B1 and the linoleic acid product arachidonic acid create the key to turn on PPAR gamma and thus the torpid metabolism.
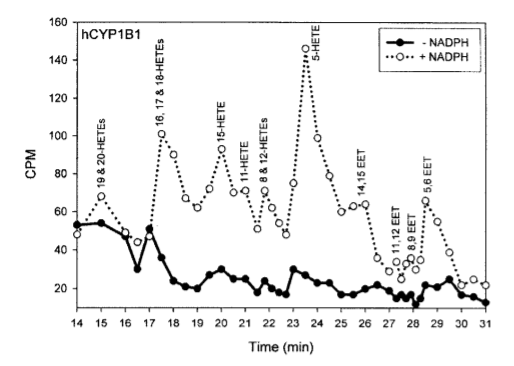
This is a lab experiment, however, is it relevant in the real world? Does it have the same effect in the liver of a living organism – the place where torpor is triggered?
Persistent Organic Pollutants
PCBs and dioxins are Persistent Organic Pollutants (POPs) – compounds with a very long halflife that persist in the environment for 100s of years. They are produced from industrial processes such as coal burning and paper manufacturing, and from tailpipe emissions and trash incineration. PCBs are famously high in the Hudson River. Dioxins have the unfortunate ability to bio-accumulate in fat tissues of animals (and humans) who consume them.
Dioxins are known activators of the AhR. If you feed dioxins to a mouse, you can see whether turning on the AhR creates these oxidized PUFA in real life.
The AhR oxidizes PUFA
This study6 looked at the effects of dioxin treated mice with or without AhR. I’m going to focus on results in the liver since that is where lipogenic genes tend to be upregulated in obesity. They found that:
- Dioxin did not increase eicosanoid (oxidized PUFA) levels in mice without AhR. Thus, the observed increases in the eicosanoids induced by dioxin are dependent on AhR.
- In the absence of dioxin, the wild type and Ahr lacking mice exhibited similar levels of eicosanoids.
- In the liver, dioxin increased the levels of several epoxides of arachidonic acid (EETs), a-linolenic acid (EpoDEs), EPA (EpETEs) and DHA (EpDPEs), in several cases more than 10-fold. Dioxin increased the levels in the liver of an even greater number of the diols of arachidonic acid, EPA and DHA (DHETs, DiHETEs and DiHDPEs, respectively) and also increased the levels of the diols of linoleic acid (DiHOMES).
So yes, The AhR creates oxidized PUFA in response to dioxin.
Blocking the AhR Lowers CYP1B1 and prevents Diet Induced Obesity in mice
When mice are fed a Western diet, they make lots of CYP1B1 and become obese. When a pharmaceutical is used that blocks the AhR, CYP1B1 expression is dramatically reduced and obesity is prevented. This is despite the fact that whether or not the AhR is blocked the mice eat the same number of calories. Obesity is not caused by overeating, it is a fuel partitioning disorder.
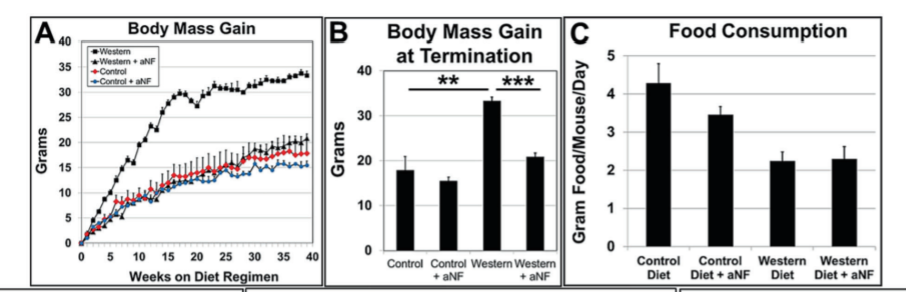
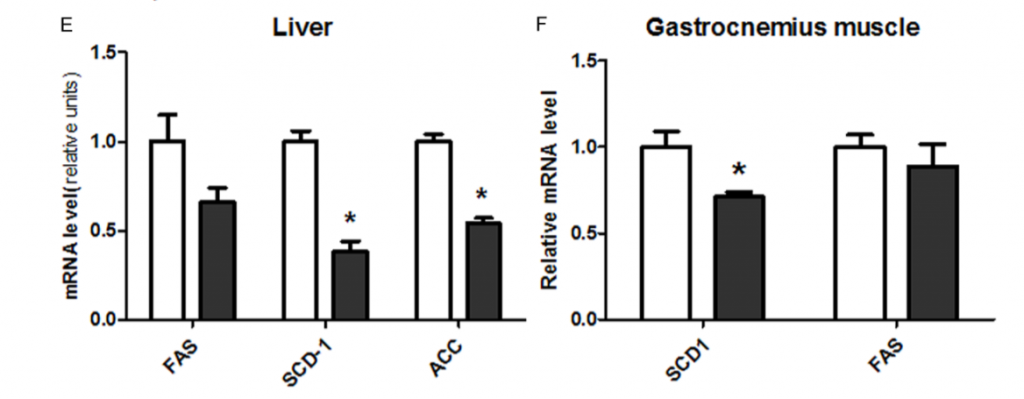
Furthermore, mice with a CYP1B1 gene knockout (they lack CYP1B1) have lower expression levels of SCD1 and the other lipogenic genes and are (somewhat) resistant to obesity.7
Evidence That the AhR/CYP1A1/CYP1A2/CYP1B1 axis affects human obesity
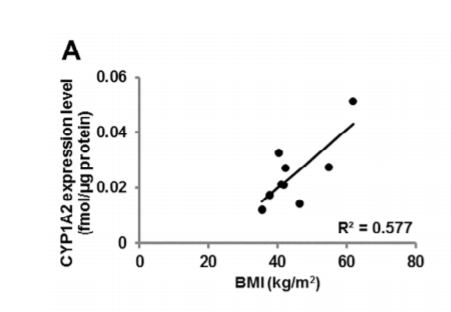
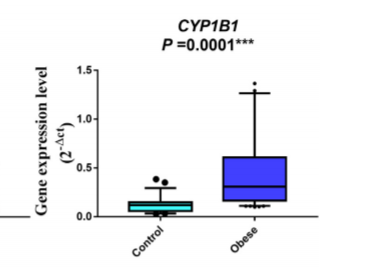
Dioxin levels are directly associated with abdominal obesity.8 AhR and CYP1B1 are highly expressed in blood cells of obese children compared to controls.9 A gene polymorphism in CYP1A1 was associated with abdominal obesity in Egyptian women.10 There was a direct correlation between BMI and intestinal levels of CYP1A2 in obese adults undergoing bariatric surgery.11
Blocking the AhR cures obesity in mice
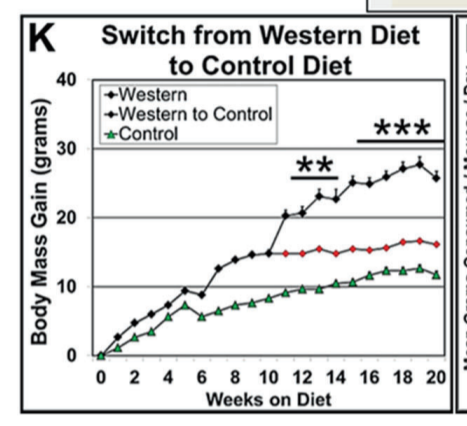
I first mentioned this in The SCD1 Theory of Obesity Part 2. This was the experiment:12
Three groups of mice were made fat on a Western diet while control (lean) mice ate regular chow food. The three groups of mice either: 1) remained on the Western diet, 2) which switched back to the control diet or 3) remained on the Western diet but with a pharmaceutical inhibitor of the AhR.
The mice kept on the Western diet had massively increased SCD1 levels and became the fattest. The mice switched to the control diet still had massively increased SCD1 levels and remained obese compared to controls. The mice with the AhR blocked on the Western diet had SCD1 levels reduced to control levels and immediately returned to the weight of lean controls.
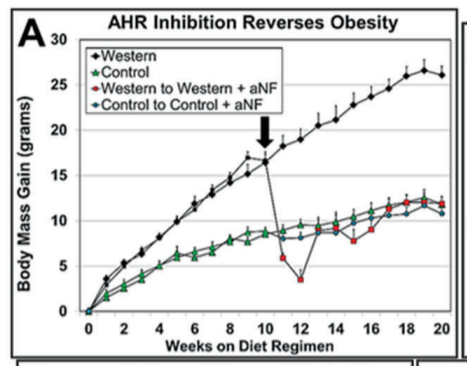
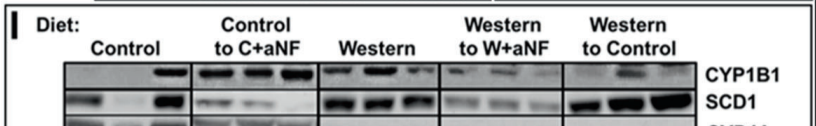
The other thing this experiment showed is that the level of an enzyme called phospholipase A2 (Pla2g4a) was greatly increased in Western Diet fed mice but that the increase was eliminated by blocking the AhR. The job of Pla2g4a is to release arachidonic acid from membranes so that it can get oxidized by CYP1B1 into keys to turn on PPAR gamma. So stimulating the AhR both mobilizes and oxidizes PUFA.
The AhR is Stimulated in the liver by oxidized LDL
When you consume PUFA, your liver packs it into particles called VLDL and releases the VLDL into the bloodstream. Cells “eat” the fat in the VLDL and as they become smaller, they are called LDL, which many of you know of as “bad cholesterol”. The LDL tend to collect PUFA and if this PUFA oxidizes the LDL becomes oxidized LDL (oxLDL). oxLDL in the bloodstream signals TLR4, a receptor in the outer membrane of liver cells, which activates the AhR through a pathway involving NF-kB and an enzyme called IDO1.13
IDO1 converts the amino acid tryptophan into something called kynurenine, which directly activates the AhR. The AhR ALSO activates IDO1, thus creating a positive feedback loop. The AhR activates IDO1 which makes kynurenine, which activates the AhR.
The Full Pathway from PUFA to torpor
You eat PUFA, your liver packs it into VLDL, which ultimately creates oxLDL. The oxLDL stimulates the AhR in your liver. The AhR upregulates the cytochrome P450 enzymes and phospholipase A2, which create the specific oxidized PUFA that is the key to turning on PPAR gamma. PPAR gamma turns on the lipogenic genes SCD1, ACC, FASN, ELOVL6 and the others. Obesity and torpor ensue.
Dealing with the AhR
There may be some good news here for humans. This 2016 paper14 suggests that a genetic change in the AhR that differentiates us from neanderthals and other primates makes the human AhR much less inducible by xenobiotics such as dioxins. The theory is that as we learned to use fire to cook our food, we adapted to constant exposure to smoke and charred meat by evolving a less active AhR. This 2020 paper15, however, refutes that finding.
Depressingly, for those of us who are not exposed to dioxins in the workplace, our main source is animal fat. The dioxins from industrial products are widespread and occur in small amounts in soil and plants and bio-accumulate in animal fat. According to the EPA, typical American consumption is 119 picograms, which they claim is a safe level.
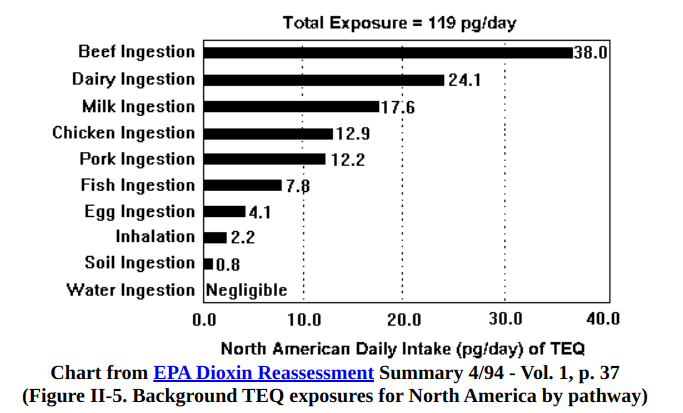
Other potential sources of compounds that would activate the AhR include charred food, cigarette smoke, auto exhaust. Basically anything that’s on fire. Probably also fryer oil.
Interestingly, resveratrol blocks the activation of the AhR.16 I’ve talked about Resveratrol on this site before as something that downregulates SCD1. Perhaps the mechanism by which this works is its suppression of AhR.
Check back in, the next article will show how to REALLY combat the effects of the AhR.
- 1.Ye W, Chen R, Chen X, et al. AhR regulates the expression of human cytochrome P450 1A1 (CYP1A1) by recruiting Sp1. FEBS J. Published online June 25, 2019:4215-4231. doi:10.1111/febs.14956
- 2.Sulem P, Gudbjartsson DF, Geller F, et al. Sequence variants at CYP1A1–CYP1A2 and AHR associate with coffee consumption. Human Molecular Genetics. Published online February 28, 2011:2071-2077. doi:10.1093/hmg/ddr086
- 3.Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. METABOLISM OF RETINOIDS AND ARACHIDONIC ACID BY HUMAN AND MOUSE CYTOCHROME P450 1B1. Drug Metab Dispos. Published online July 16, 2004:840-847. doi:10.1124/dmd.32.8.840
- 4.Chen GG, Xu H, Lee JFY, et al. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cellsvia a peroxisome proliferator-activated receptor gamma-dependent pathway. Int J Cancer. Published online October 14, 2003:837-843. doi:10.1002/ijc.11447
- 5.HAN J, SUN L, XU Y, LIANG H, CHENG Y. Activation of PPARγ by 12/15-lipoxygenase during cerebral ischemia-reperfusion injury. International Journal of Molecular Medicine. Published online November 12, 2014:195-201. doi:10.3892/ijmm.2014.1998
- 6.Brulport A, Le Corre L, Chagnon M-C. Chronic exposure of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces an obesogenic effect in C57BL/6J mice fed a high fat diet. Toxicology. Published online September 2017:43-52. doi:10.1016/j.tox.2017.07.017
- 7.Liu X, Huang T, Li L, et al. CYP1B1 deficiency ameliorates obesity and glucose intolerance induced by high fat diet in adult C57BL/6J mice. Am J Transl Res. 2015;7(4):761-771. https://www.ncbi.nlm.nih.gov/pubmed/26064443
- 8.Chang J-W, Chen H-L, Su H-J, Lee C-C. Abdominal Obesity and Insulin Resistance in People Exposed to Moderate-to-High Levels of Dioxin. Folli F, ed. PLoS ONE. Published online January 11, 2016:e0145818. doi:10.1371/journal.pone.0145818
- 9.Shahin NN, Abd-Elwahab GT, Tawfiq AA, Abdelgawad HM. Potential role of aryl hydrocarbon receptor signaling in childhood obesity. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids. Published online August 2020:158714. doi:10.1016/j.bbalip.2020.158714
- 10.Bayoumy N, El-Shabrawi M, Younes S, Atwa K. CYP1A1 gene (6235T<C) polymorphism as a risk factor for polycystic ovarian syndrome among Egyptian women. Human Fertility. Published online October 22, 2018:142-147. doi:10.1080/14647273.2018.1522455
- 11.Miyauchi E, Tachikawa M, Declèves X, et al. Quantitative Atlas of Cytochrome P450, UDP-Glucuronosyltransferase, and Transporter Proteins in Jejunum of Morbidly Obese Subjects. Mol Pharmaceutics. Published online July 19, 2016:2631-2640. doi:10.1021/acs.molpharmaceut.6b00085
- 12.Rojas IY, Moyer BJ, Ringelberg CS, Tomlinson CR. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of CYP1B1, PPARα, SCD1, and osteopontin. Int J Obes. Published online January 7, 2020:948-963. doi:10.1038/s41366-019-0512-z
- 13.Moyer BJ, Rojas IY, Kerley-Hamilton JS, et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicology and Applied Pharmacology. Published online June 2016:13-24. doi:10.1016/j.taap.2016.03.011
- 14.Hubbard TD, Murray IA, Bisson WH, et al. Divergent Ah Receptor Ligand Selectivity during Hominin Evolution. Mol Biol Evol. Published online August 2, 2016:2648-2658. doi:10.1093/molbev/msw143
- 15.Aarts JMMJG, Alink GM, Franssen HJ, Roebroeks W. Evolution of Hominin Detoxification: Neanderthal and Modern Human Ah Receptor Respond Similarly to TCDD. Mulligan C, ed. Molecular Biology and Evolution. Published online November 24, 2020:1292-1305. doi:10.1093/molbev/msaa287
- 16.Assefa EG, Yan Q, Gezahegn SB, et al. Role of Resveratrol on Indoxyl Sulfate-Induced Endothelial Hyperpermeability via Aryl Hydrocarbon Receptor (AHR)/Src-Dependent Pathway. Oxidative Medicine and Cellular Longevity. Published online November 27, 2019:1-15. doi:10.1155/2019/5847040
Thank you, Brad Marshall.
You’re welcome!
So what data do you have on the timeline of dioxins and/ or other herbicides introduced into environment?
Fibrates used as drugs to control cholesterol are herbicides and induce CP450 to oxidize PUFA and upregulated soluble epoxide hydrolase…..imcrease in inflammation. However studies tend to show decreased heart disease.
I don’t know the exact timeline but I’d guess increasingly as the 20th century went on. A friend grew up in a paper mill town in Maine in the 80’s. Notorious for dioxins and both of their parents had cancer.
Don’t know if you want to go down this rabbit hole but I’ve been looking into claims that Vitamin A isn’t really a vitamin and is acting as a toxin which we evolved to be able to handle but with environmental and dietary changes in our society we no longer have the ability to regulate it and are also exposed to unnaturally high levels. It sounds absurd at first but if you really delve into it there are some interesting findings. The fellow that posited this issue has been in an extremely low Vitamin A diet for 7 years now and has only seen improvement in his health with no signs of the deficiency that would be expected. One of his observations is the effects on the liver and he notes a connection with obesity. Just something else to think about. https://ggenereux.blog/
I’m working on the followup article. The thing is that the dimer partner of PPAR gamma is the “retinoid X receptor” – activated by vitamin A and required for PPAR gamma signalling. I’m not ready to call vitamin A a toxin, but it does play a role in this.
I certainly hope not. I eat a lot of beef liver crisps. 1 serving has 3710mg of Vit. A or 410% RDA. Hopefully there is a differentiation between the fully formed Vit A found in animal products vs the plant form carotenoids that have to be converted by the body.
So to sum up…
Obesity is not caused by overeating, It’s a fuel partitioning disorder. It’s largely driven in the modern Western diet by polyunsaturated fatty acids (PUFA) which are the primary kind of fat in soy, corn, and canola oil, consumed with carbohydrates. That combination occurs in virtually all processed foods.
That combination puts the body in the state of TORPOR, which is essentially pre-hibernation when the body is signaled to store rather than burn fat.
Is that a reasonable summary?
I’ll let Brad confirm but that sounds right to me. Underpinning this is an interesting (new?) field of nutrition whose name should be chosen- the science of informing macro-physiological stories to the body using nutrient ratios & presence.
That’s a perfect name with a great acronym: TSOIMPSTTBUNRP. We may have to work on it 😉
We can shorten it to FATASSBEAR for the listeners!
Yeah, that’s pretty much it!
I refuse to believe that a croissant is a processed food. But it is relatively poor in PUFA.
I’m not ready to swallow that antagonizing PPAR gamma is a good thing.
https://onlinelibrary.wiley.com/doi/abs/10.1111/jgh.15025#:~:text=The%20PPAR-%CE%B3%20agonist%20made%20lipid-induced%20M1-polarized%20macrophages%20switch,shifting%20subsequently%20affected%20lipid%20metabolism%20in%20primary%20hepatocytes.
It depends on where you antagonize it. Activated PPARy in the liver makes you fat quickly. ACtivated PPARy in fat cells makes you thermogenic over time.
Any thoughts on this?
https://pubmed.ncbi.nlm.nih.gov/17082484/
“These findings suggest that saturated FAs, which are released in large quantities from hypertrophied adipocytes via the macrophage-induced adipocyte lipolysis, serve as a naturally occurring ligand for TLR4, thereby inducing the inflammatory changes in both adipocytes and macrophages through NF-kappaB activation.”
Far as I can tell from skimming the full-text link, the “saturated fatty acid” they’re talking about is Palmitic acid, as I don’t see any references to Stearic.
This is interesting to me because the idea that palmitate – which can be produced endogenously via DNL – may trigger torpor seems consistent, yet it poses a stark problem with feeding. Does Palm Oil induce torpor?
Palm oil makes mice very fat.
That is interesting, although I heed Peter’s caveat:
“The high palmitic acid gives the most weight gain as it delivers 4.5% of calories as PUFA. Olive oil is a close second, also with 4.5% linoleic acid. The oddity is the safflower oil diet which is very high in PUFA but only gives intermediate obesity.”
Either way this study is awesome because it answers the next problem I was going to ask about:
Does the Palmitic acid TLR4 effect (purportedly existing per the pubmed article I posted) sabotage high-stearic acid animal or plant fats’ ability to avoid torpor, the answer is no as demonstrated in that study despite palmitate making up 25% of cocoa butter.