
In a recent post I talked about three groups of pigs with differing levels of insulin sensitivity.
Group 1 was fed a low fat diet and remained insulin sensitive.
Group 2 was given 40% of calories as a special soybean oil blend with the triply unsaturated alpha-linolenic acid (ALA) largely removed. These pigs became what Peter from hyperlipid would call “pathologically insulin sensitive” based on the fact that they had low fasting blood glucose in the context of very low insulin levels.
Group 3 was given regular soybean oil including the oxidation prone ALA. This group developed pathological insulin resistance as determined by a high fasting blood glucose level in the context of massively higher circulating insulin. In this group, lipogenic genes were switched on and oleic acid – the end product of lipogenesis – replaced the linoleic acid in their plasma fat.
| Pigs | Lo Fat | Soy Oil No ALA | Soy Oil |
| Blood Glucose | 8.8 | 7.8 | 9.6 |
| Insulin | 6.9 | 1.7 | 30.9 |
| DI1600 | 5.4 | 6.1 | 7.9 |
| Linoleic Acid | 14.3 | 25.8 | 11.6 |
| Oleic Acid | 35.4 | 31.5 | 38.8 |
| Weight Gained (kg) | 24.4 | 25.1 | 26.6 |
In a follow up article I said that pigs who remained lean on a high PUFA diet had the super power of linoleic acid accumulation. Somehow they can accumulate linoleic acid forever without turning on lipogenic genes and the torpid metabolism.
PPAR gamma is the gatekeeper of the torpid metabolism
This 2018 paper demonstrates that it is PPAR gamma that controls the switch into a torpid metabolism1. Obesity prone mice were given either a low fat or a high fat diet – 60% of calories as a high-PUFA lard and soybean oil, with protein from purified casein with some sugar and purified starch. This particular high fat diet is known to cause obesity and insulin resistance quite quickly in this strain of mice.
The mice were further divided into groups with normal PPAR gamma levels or mice whose PPAR gamma activity had been eliminated. The mice on the high fat diet who lacked PPAR gamma look like the pigs who were given the soybean oil with the ALA removed – they are very insulin sensitive! The mice with functional PPAR gamma become pathologically insulin resistant. Lack of PPAR gamma prevents insulin resistance.
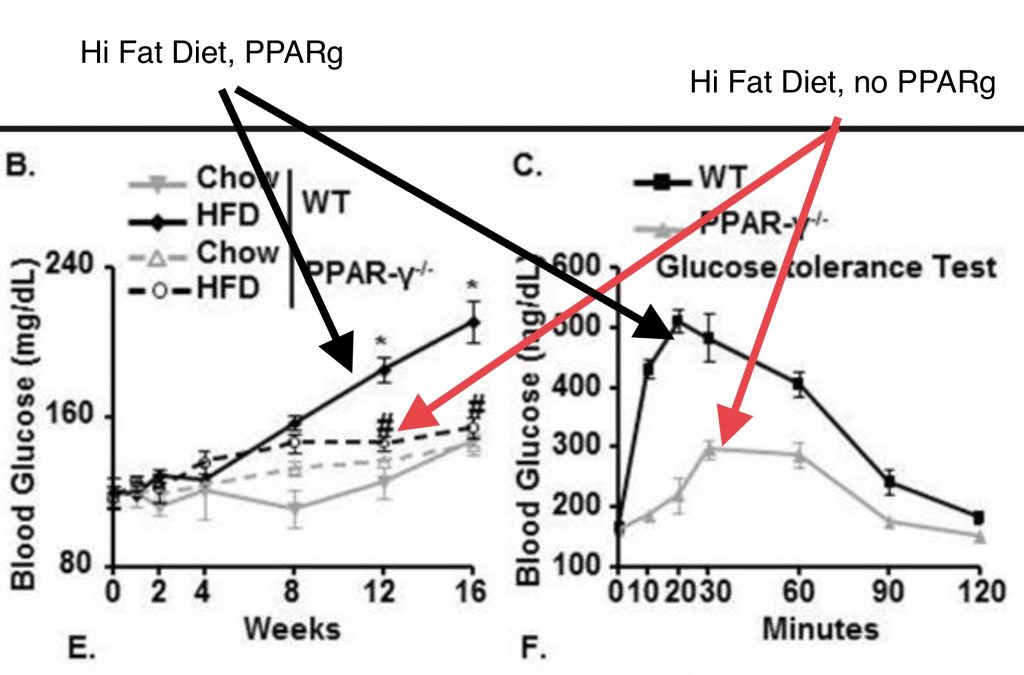
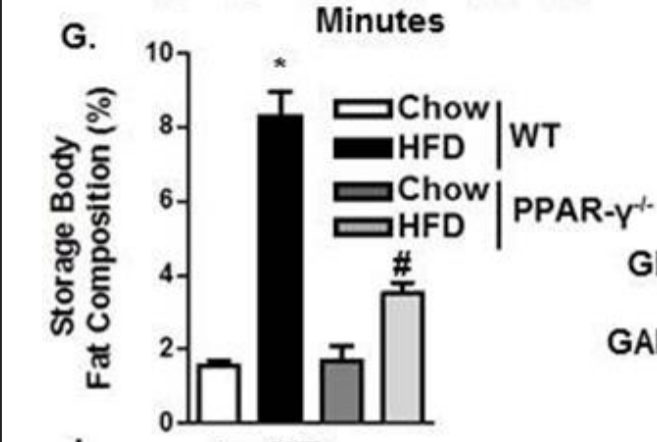
Mice lacking PPAR gamma also gained significantly less weight than “normal” mice on the high fat diet, just like the pigs with the ALA removed soybean oil.
I’ve talked about PPAR gamma before, the “Master Regulator of Adipogenesis and Obesity”.2 It is a transcription factor that turns on other genes, including the lipogenic genes such as SCD1. It may be beneficial when it is activated in fat cells, leading to the creation of thermogenic brown and beige fat cells. When it is active in the liver and muscle tissue you will store fat.
PPAR gamma is activated by oxidized PUFA
PPAR gamma is “ligand activated”, which means that it is only turned on when it binds to a certain target (AKA ligand). Specifically, PPAR gamma binds to certain oxidized PUFA. For instance, PPAR gamma is activated by 15-HETE3, an oxidized linoleic acid metabolite (OXLAM)4. PPAR gamma has a non-specific ligand binding pocket, however. It can bind to a variety of ligands, many of which have presumably not been characterized yet.
Let’s think again about the pigs given regular soybean oil. The presence of ALA was the difference between highly insulin sensitive pigs and very insulin resistant pigs. ALA is an omega 3 fatty acid with three double bonds that is highly prone to oxidation. The reason that flaxseed oil is sold in a dark brown bottle is because it’s mostly ALA. If light hits flaxseed oil it oxidizes into linseed oil: wood polish.
So the presence of ALA presumably provided an oxidized metabolite which switched on PPAR gamma in the pigs which in turn turned on the lipogenic genes and made them torpid.
Back to the mice. ALA isn’t necessary for torpor, but it may get you there a bit quicker. The mice given the high fat diet had plenty of linoleic acid and were able to become torpid just fine. Until the PPAR gamma was removed. Without PPAR gamma, insulin sensitivity was maintained.
PPAR gamma activity in the liver and muscle tissues is the gatekeeper into torpor.
PDK4 and Metabolic Inflexibility
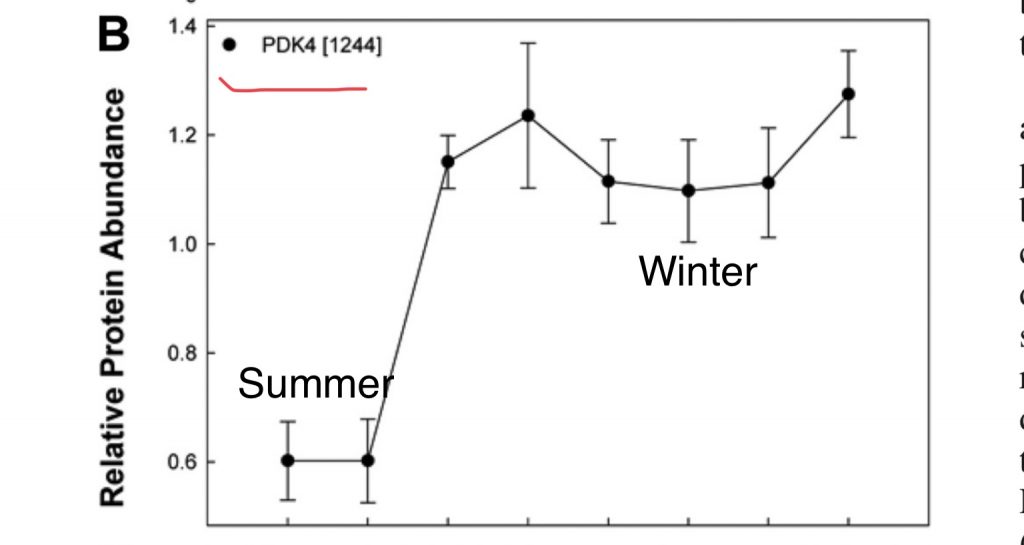
How does PPAR gamma make a mouse insulin resistant? The authors of the paper go on to show that PPAR gamma binds to the promoter of a gene called PDK4 and turns it on. The torpid, insulin resistant mice have the highest levels of PDK4 and the highest blood glucose levels.

This is interesting because obese and diabetic humans have high levels of PDK4 expression in skeletal muscle compared to lean humans.5
| PDK4 Expression | Low Fat Diet | High Fat Diet |
| Lean Human Muscle | 0.16 | 0.23 |
| Obese Human Muscle | 0.31 | 0.93 |
Even more interesting is the fact that a major change between torpid hibernators in fall and winter versus summer is that in winter they massively up-regulate PDK4 in skeletal muscle.6 So once again, we see a parallel between a torpid hibernator, an obese human, pigs fed soybean oil and an obesity prone mouse on a high fat diet. We don’t KNOW that the pigs have higher PDK4 levels because the authors didn’t look into it, but I would put some money on it and I don’t even enjoy betting.
Here’s what PDK4 does. When glucose is taken up by a cell, it is converted to pyruvate in the cytosol (cell water) by a process called glycolysis. Pyruvate enters the mitochondria and the pyruvate dehydrogenase complex (PDC) converts it to Acetyl-CoA where it is burned as fuel. PDK4 (pyruvate dehydrogenase kinase 4) phosphorylates the PDC and turns it OFF. When PDK4 is at high levels, the PDC is inactive and glucose cannot be efficiently burned.
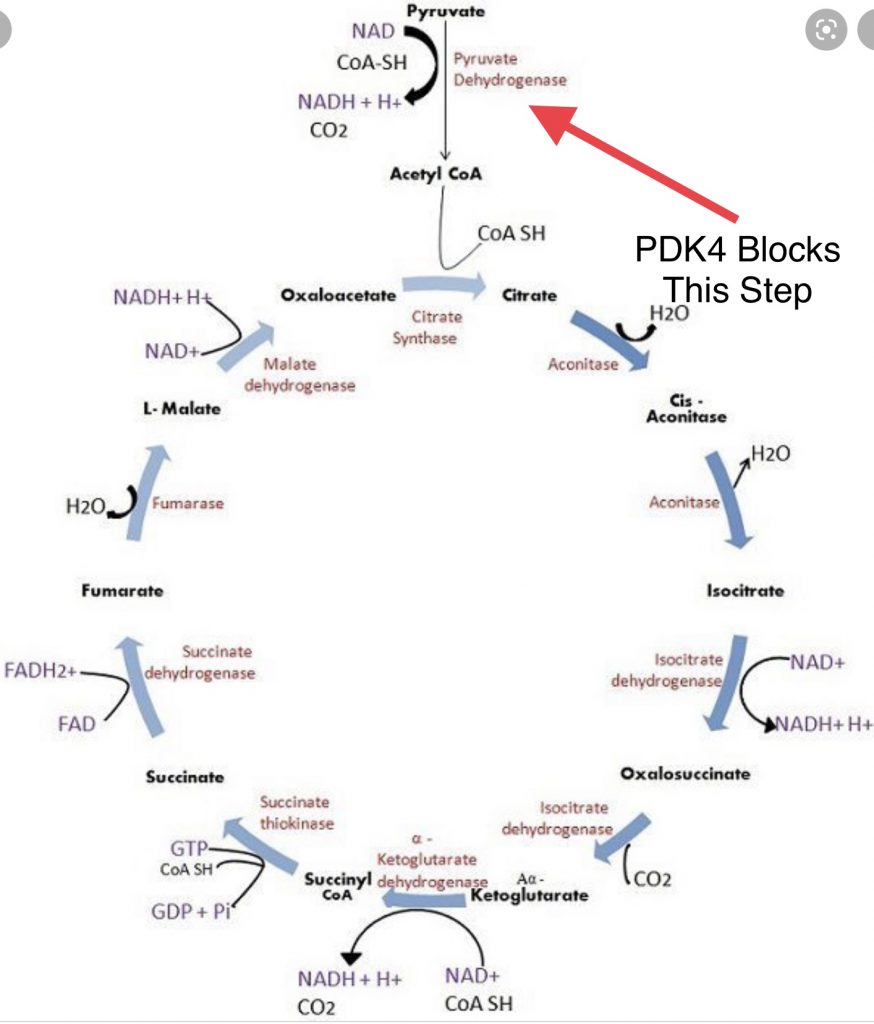
This makes sense if you’re a hibernating animal planning to live on your stored fat all winter. A hibernating bear lives off of stored body fat all winter. There is no source of starch. High PDK4 expression keeps the bear set to burn fat.
A healthy omnivore, however, should be able to use glucose efficiently when it is available. One of the main differences between obese and lean humans is that obese humans burn glucose less efficiently. They are making too much PDK4 in muscle tissue.
As quantitatively the largest organ in the body, skeletal muscle accounts for 30% to 40% of the metabolic rate in adults in the resting state. Contributing to 80% of insulin-stimulated glucose uptake, it is a major site for glucose oxidation and fatty acid metabolism. The skeletal muscle exhibits remarkable metabolic flexibility in fuel usage in response to various metabolic challenges such as energy deprivation and changes in diet composition. In lean healthy individuals, under insulin stimulation, skeletal muscle is able to switch from predominantly lipid oxidation and high rates of fatty acid uptake to the suppression of lipid catabolism and elevated glucose uptake, oxidation and storage. However, obese and T2D patients manifest greater rates of lipid oxidation in skeletal muscle and are relatively insulin resistant, resulting in metabolic inflexibility.
Zhang, et al. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility
They are making too much PDK4 because they have oxidized PUFA that switched on PPAR gamma in their muscle tissue. They are metabolically inflexible because of the PDK4.
- 1.Sikder K, Shukla SK, Patel N, Singh H, Rafiq K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-γ. Cell Physiol Biochem. Published online 2018:1317-1331. doi:10.1159/000492091
- 2.Shao X, Wang M, Wei X, et al. Peroxisome Proliferator-Activated Receptor-γ: Master Regulator of Adipogenesis and Obesity. CSCR. Published online March 3, 2016:282-289. doi:10.2174/1574888×10666150528144905
- 3.Chen GG, Xu H, Lee JFY, et al. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cellsvia a peroxisome proliferator-activated receptor gamma-dependent pathway. Int J Cancer. Published online October 14, 2003:837-843. doi:10.1002/ijc.11447
- 4.Green FA. Transformations of 5-HETE by activated keratinocyte 15-lipoxygenase and the activation mechanism. Lipids. Published online October 1990:618-623. doi:10.1007/bf02536012
- 5.Bergouignan A, Gozansky WS, Barry DW, et al. Increasing Dietary Fat Elicits Similar Changes in Fat Oxidation and Markers of Muscle Oxidative Capacity in Lean and Obese Humans. Blanc S, ed. PLoS ONE. Published online January 12, 2012:e30164. doi:10.1371/journal.pone.0030164
- 6.Hindle AG, Karimpour-Fard A, Epperson LE, Hunter LE, Martin SL. Skeletal muscle proteomics: carbohydrate metabolism oscillates with seasonal and torpor-arousal physiology of hibernation. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. Published online November 2011:R1440-R1452. doi:10.1152/ajpregu.00298.2011
Maybe coffee polyphenols reduce PDK4?
https://journals.physiology.org/doi/full/10.1152/ajpendo.00441.2010
Interesting find. Sounds like it!
Nice, Rachel.
I think you meant if light hits flaxseed oil it will oxidize into linseed oil.
Thanks, I’ve had that editor fired.
I am a bit confused with this post.
Doesn’t PPAR gamma increase insulin sensitivity by shuffling fatty acids into fat cells, thereby encouraging cells to utilize glucose?
I tried investigating this more and everything I am finding indicates that PPAR alpha upregulates PDK4 and PPAR gamma downregulates it. For example:
https://pubmed.ncbi.nlm.nih.gov/15955060/
“PPARalpha and PPARdelta agonists were found to specifically upregulate PDK4 mRNA expression, whereas PPARgamma activation selectively decreased PDK2 mRNA transcript abundance.“
Or from
https://www.researchgate.net/publication/11276124_Up-regulation_of_pyruvate_dehydrogenase_kinase_isoform_4_PDK4_protein_expression_in_oxidative_skeletal_muscle_does_not_require_the_obligatory_participation_of_peroxisome-proliferator-activated_recepto
“Signaling via peroxisome-proliferator-activated receptor alpha (PPARalpha) is an important component of the mechanism enhancing hepatic and renal PDK4 protein expression. Activation of PPARalpha in gastrocnemius, a predominantly fast glycolytic (FG) muscle, also increases PDK4 expression, an effect that, if extended to all muscles, would be predicted to drastically restrict whole-body glucose disposal.”
What am I missing here?
This is what makes biology so fun!
I think what we’re seeing is the difference between the short and long term regulation of genes. PPAR alpha increases PDK4 accutely after meals. This is normal. After your blood glucose drops, PPAR alpha and PDK4 increase and the proportion of fat that is burned is increased.
Long term is different. There is another regulator of PDK4: miRNA-182. I thought about putting in this post but opted not to because I wanted to keep it reasonably simple.
miRNA-182 is increased in response to ROS and it blocks production of PDK4 protein. PPAR gamma in the long term upregulates SCD1, which lowers the activity of mitochondrial Complex II, which limits ROS production. miRNA-182 levels drop and PDK4 increases.
Brad
Curious whether the negative effects of ALA might also be produced by the other omega 3s – EPA and DHA – when we eat foods like fish? Or is that effect unique to ALA among the omega 3s?
I suspect there is a U-shaped curve involved here. A little bit of long chain Omega-3 in the context of a relatively low-PUFA diet seems to be beneficial in the sense of lowering expression of lipogenic genes and SCD1. On the other hand, bears fatten up for winter by eating salmon.
Brad
Ray peat strikes again
Whatever happened to the PPAR alpha?
I did not read that you were still suggesting PPAR gamma inhibition.
My understanding of his writings so far is that PPAR gamma is really causing a lot of trouble and it’s linoleic acid largely responsible for triggering it..
PPAR alpha could probably use some refreshers for those of us following along. My understanding is it’s the other side of the knife for “getting out of” torpor, promoting the burning down of fat, but I am curious in lieu of these details what PPAR alpha does under the torpor metabolic state.
In the next few weeks we’ll introduce a few more of the “nuclear recpeter superfamily”, ehich includes all three PPARs and look at how they work together.
Brad
What about DHA/epa vs ALA ?
Probably good in small amounts/bad in large amounts.
What that means downstream is that uptake of glucose will be almost impossible: “The impairment of glucose metabolism (. . . ) is mediated by the short-term inhibition of several glycolytic processes. The extent of inhibition increases along the glycolytic pathway, being most severe at the level of pyruvate dehydrogenase and less severe at the level of glucose uptake and PFK-1. (. . . ) It has been proposed that these changes lead to an accumulation of cytosolic citrate, which in turn inhibits PFK-1, followed by an increase in glucose-6-phosphate, which eventually inhibits hexokinase.”
While this argument from the english wikipedia (“Randle cycle”) refers to the condition of fat burning in the mitochondria, it will be the same with PDK 4, I guess. Only long-term! Explanatory value for insulin resistance?! – Pedro, Spain