
In part I of this series I introduced the analogy of reductive stress as a flooded engine. If there is too much fuel and not enough air, an engine will flood. Elevated circulating acylcarnitines in obesity are analogous to black smoke out of the tailpipe of a flooded engine – unburnt fuel spewing back out of the pistons.
My ATV in the late 1980’s had a carburetor – a device that mixes the fuel with air before it is sent to the pistons. Your car has digital fuel injection, so flooding the engine isn’t a problem. A useful analogy that will be lost on future generations.
The ATV had a carburetor screw that adjusted the air-fuel mix. The bulk of the airflow was independent of the screw. While the ATV was running you could open the screw or close it all of the way and it would still run. But if you adjusted the screw to where the fuel mix was too rich, it would run rough and you’d get the black smoke.
The Mitochondrial carburetor
The mitochondria also mixes fuel (electrons from hydrocarbon bonds) with oxygen, just like a carburetor. In the mitochondria, NAD+ takes the place of oxygen in the pistons, oxidizing the fuel and carrying the electrons to the electron transport chain (ETC). The electron transport chain uses the energy of the electrons to generate ATP and the electrons recombine with oxygen as they come out of the ETC at complex IV. This is the classic view and this is the bulk of the oxygen available for burning fat.
Except that the high energy electrons ALSO combine with oxygen at complexes I and III, forming superoxide. This extra bit of oxygen injected into the system is the carburetor screw making subtle adjustments to the air fuel mixture to ensure that fat is burned completely and that there is no black smoke. Here is the more nuanced view.1
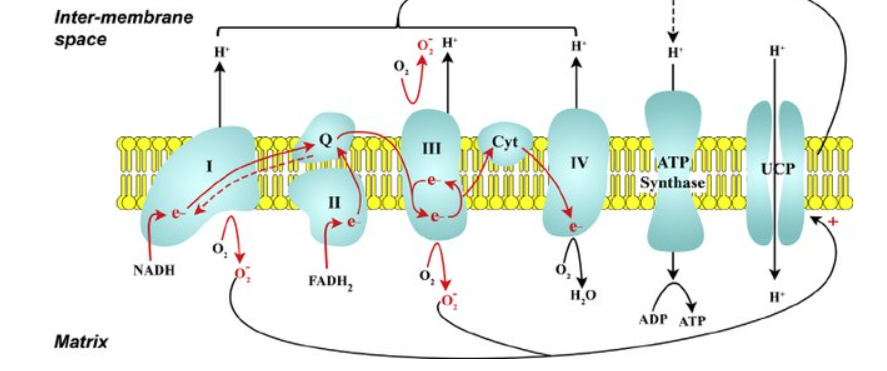
The reverse dotted arrow from coenzyme Q indicates the mitochondrial bottleneck. Complex I and complex II both have to pass electrons to coenzyme Q. If there is high input through both complex I and II, the electrons move backwards through the bottleneck to create much more superoxide, bringing more oxygen into the air fuel mix.
There are two inputs to complex II. The first is succinate being oxidized to fumarate by succinate dehydrogenase in the TCA cycle, which every molecule of acetyl-CoA can generate. The second is the enzyme Acyl-CoA Dehydrogenase. Acyl-CoA Dehydrogenase puts a double bond into a fat molecule as the molecule is broken down into acetyl-CoA and inputs the electrons into the ETC at complex II (FAD).
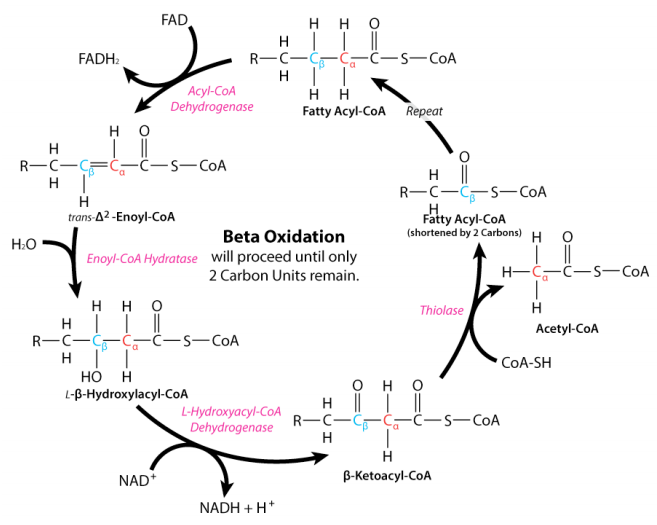
IF THERE IS ALREADY A DOUBLE BOND THERE IS NO NEED FOR ACYL-COA DEHYDROGENASE!! Unsaturated fats have less inputs to FAD (complex II) and therefore drive less superoxide production at complex I and burn with a richer air-fuel mix (less oxygen). They burn dirty.
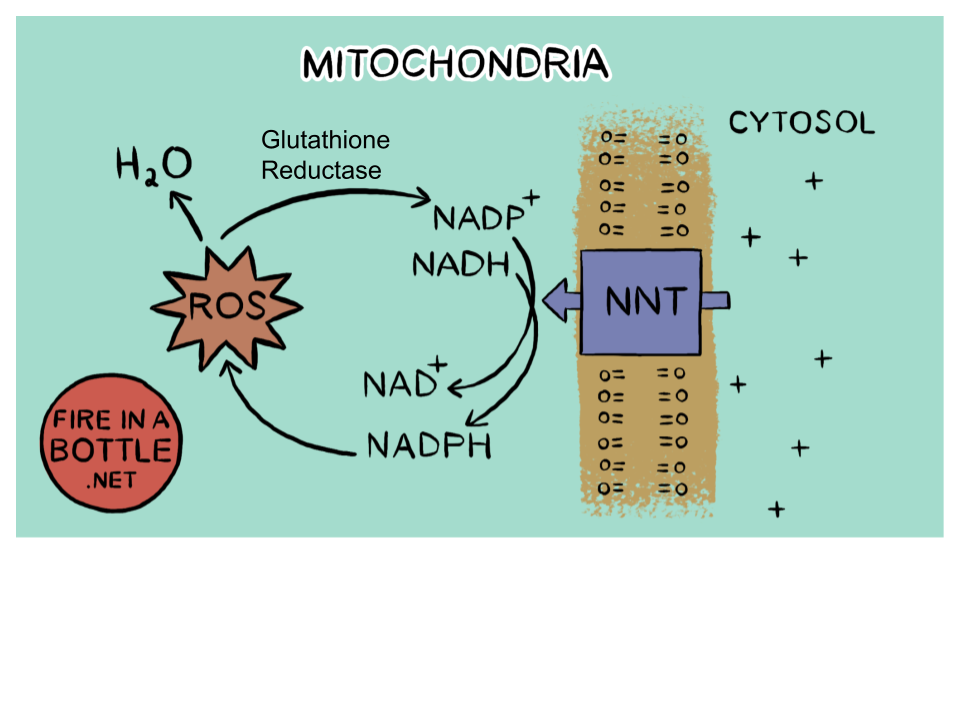
Superoxide/Glutathione Reductase/NNT turns superoxide into NAD+
The mitochondria (of course) has a very efficient system for converting the oxidizing power of superoxide to NAD+. The ETC isn’t set up like this by accident, it was chosen by evolution to be the best system. The mitochondrial bottleneck is the carburetor screw that ensures that when saturated fat is burning, the fuel is properly oxygenated.
If this all sounds a bit theoretical, consider what happens to NAD+/NADH ratio when cells in culture are given pure palmitate (PA – saturated fat) versus a mix of palmitate and oleic acid (OA – MUFA)2. It’s not subtle.
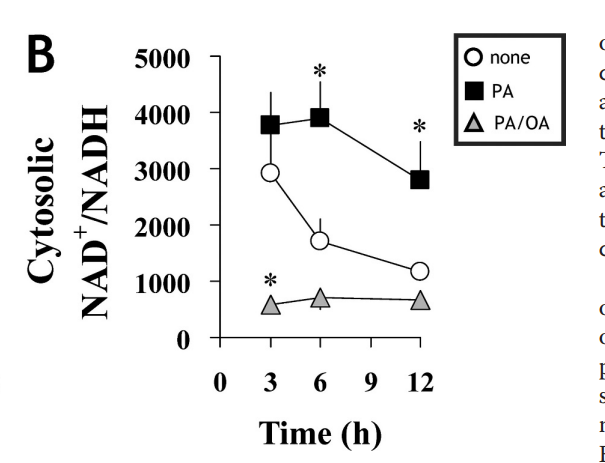
The palmitate drives a huge amount of superoxide/NAD+. Probably too much in fact, to the point of legit oxidative stress. The good news is that pure long chain saturated fat never occurs in nature, so you’ll never have to worry about this. This experiment adds to the evidence from mice lacking SCD1, who cannot make MUFA, who have very saturated fat, who have high levels of NAD+, who are hypermetabolic and resistant to obesity. A high ratio of NAD+/NADH ensures that you’re not burning dirty.3
Monounsaturated fat (MUFA) is unsaturated enough to bring the NAD+ levels to below baseline. PUFA, of course, has even less input to the bottleneck and therefore burns less oxygen. Pufa burns very rich (dirty).
Chipmunks
Most authors who have written about superoxide production at complex I consider it “bad” but a sort-of-inevitable consequence of oxidative biology. If you assume the system is designed this way on purpose, a lot of questions about obesity become easier to answer. If we consider superoxide generation to control the air-fuel mixture, suddenly eating PUFA seems like a bad idea. Won’t that burn dirty?
It will burn dirty. That is why the chipmunks need it to get their mitochondrial enzymes fully acetylated. Multiple studies4–7 have shown that hibernating animals require significant PUFA in their diets to become fully torpid. They need that dirty burning fuel to drive a buildup of acetyl-CoA – the source of the acetyl groups – in their mitochondria. Acetylated mitochondrial enzymes are what turn them off and allow the chipmunks to get fully torpid. The saturated fat of sheep suet leads to consistently higher metabolic rate and oxygen consumption.
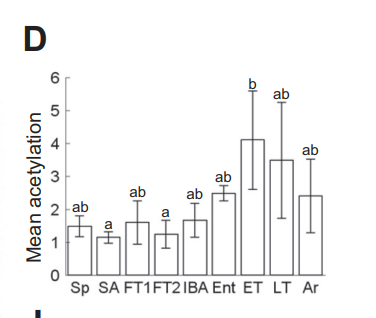
Once again, let’s look at the acetylation of mitochondrial enzymes as a hibernator cycles through torpor.8 There is a huge rise in acetylation during entrance to torpor (ENT) and early torpor (ET) that quickly recedes when animals want to come out of torpor (Ar).
Humans and PUFA
Looked at from this perspective, human obesity isn’t a pathology. We have made a biological decision. We have chosen to use a fuel that burns dirty and puts us into a torpid state. Of course, if that were true, we’d expect to see increased acetylation in obesity. To quote from a recent review on this topic9:
In particular, lysine acetylation of non-histone proteins, which controls a diverse family of mitochondrial metabolic pathways, contributes to the cardiac energy derangements seen in obesity, diabetes, and heart failure. Lysine acetylation is controlled both via acetyltransferases and deacetylases (sirtuins), as well as by non-enzymatic lysine acetylation due to increased acetyl CoA pool size or dysregulated nicotinamide adenine dinucleotide (NAD+) metabolism (which stimulates sirtuin activity).
Arata Fukushima, Gary D. Lopaschuk, Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure
Kind of like in torpor.
What Can I do?
A great first step is to avoid seed oils – including soybean, corn, sunflower, canola and safflower oils, which are found in almost all processed foods and restaurant foods. If you are obese, you may also want to avoid olive and avocado oil – you’ve already seen what MUFA can do to NAD+ levels.
If you are obese, your body fat is loaded with MUFA – the primary end product of de novo lipogenesis (DNL – making fat) – as well as the PUFA which triggered the DNL. You may need to replace some complex II activity.
In the Fire In A Bottle store, we offer several products to increase electron flow through complex II and increase mitochondrial oxidation levels. The most potent is succinade, a drinkable form of succinate, which drops its electrons straight into succinate dehydrogenase.
Pure stearic acid acts similarly to the pure palmitate that drove high NAD+ levels in the experiment above. Lastly, Sterculia Oil is from a naturally occurring tropical nut that blocks the enzymatic activity of the enzyme that converts saturated fat in your body to monounsaturated fat (SCD1).
Personally, I would recommend combining any of the above with alpha lipoic acid. Your mileage may vary.
- 1.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biology. Published online December 2015:524-551. doi:10.1016/j.redox.2015.08.020
- 2.Noguchi Y, Young JD, Aleman JO, Hansen ME, Kelleher JK, Stephanopoulos G. Effect of Anaplerotic Fluxes and Amino Acid Availability on Hepatic Lipoapoptosis. Journal of Biological Chemistry. Published online November 2009:33425-33436. doi:10.1074/jbc.m109.049478
- 3.Akie TE, Liu L, Nam M, Lei S, Cooper MP. OXPHOS-Mediated Induction of NAD+ Promotes Complete Oxidation of Fatty Acids and Interdicts Non-Alcoholic Fatty Liver Disease. Aspichueta P, ed. PLoS ONE. Published online May 1, 2015:e0125617. doi:10.1371/journal.pone.0125617
- 4.Geiser F, Kenagy GJ. Polyunsaturated lipid diet lengthens torpor and reduces body temperature in a hibernator. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. Published online May 1, 1987:R897-R901. doi:10.1152/ajpregu.1987.252.5.r897
- 5.Geiser F, Stahl B, Learmonth RP. The Effect of Dietary Fatty Acids on the Pattern of Torpor in a Marsupial. Physiological Zoology. Published online November 1992:1236-1245. doi:10.1086/physzool.65.6.30158277
- 6.Geiser F. The effect of unsaturated and saturated dietary lipids on the pattern of daily torpor and the fatty acid composition of tissues and membranes of the deer mouse Peromyscus maniculatus. J Comp Physiol B. Published online December 1991:590-597. doi:10.1007/bf00260749
- 7.Munro D, Thomas DW. The role of polyunsaturated fatty acids in the expression of torpor by mammals: a review. Zoology. Published online March 2004:29-48. doi:10.1016/j.zool.2003.12.001
- 8.Mathers KE, Staples JF. Differential posttranslational modification of mitochondrial enzymes corresponds with metabolic suppression during hibernation. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. Published online August 1, 2019:R262-R269. doi:10.1152/ajpregu.00052.2019
- 9.Fukushima A, Lopaschuk GD. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. Published online December 2016:2211-2220. doi:10.1016/j.bbadis.2016.07.020
Very interesting!
An engineers comment now: if you’re engine’s running too rich and you don’t want or can’t change air-fuel ratio, then one way is to boost your air intake by way of installing a turbo or intake blower.
The Wim Hof breathing technique explains how to improve breathing efficiency so that “exhaust” is better expelled from the body. Some of their assertions seem to lean towards fact that much of weight loss actually occurs in the lungs!
Now someone has to find a way to measure boost pressure in the mitochondria!!!
I really enjoyed reading your engine analogy! Thanks.
Well this makes sense to me. I have said for years, after people complain about digestive issues and seeds, that seeds are baby trees and plants. They don’t want to be munched on. They have toxins in them because they want to survive and grow into adults. Stay away from them.
Man-made assumptions like ROS = bad always gets us into trouble. The only correct starting point is assuming evolution is deliberate, no matter how confusing!
Yes!
The other angle is NAD consumption by inflammation or other stimulation os CD38 upregulation. In addition to lowering PUFA maybe go easy on dairy for a while to limit LPS.
Perhaps, if you are one who has an inflammatory response to dairy. Perhaps a more general statement would be to avoid things that give you inflammation personally, insomuch as you can know.
Brad, great work, I’m only layman but I still miss involvement of DECR enzyme, sorry. This I think could be the right bottleneck if PUFAs are involved. Knocking down of it can elevate acylcarnitine manytimes (C10:2 44x, C16:0 9x) and it’s function is really crutial. Your engine analogy is perfect, if NAD+ is limiting factor and NNT is the way to get it, it has to be plenty of NADP+ for conversion, but it isn’t. NADP+ is immediately converted to NADPH if glucose is available, except in liver, where serin is used. And how you can get NADP+ for NNT? I think by DECR, by PUFA beta oxidation. This bypass the normal way through superoxide (aconitase controlling signal), SOD, H2O2 (enzyme controlling signal), glutathion, NADP+. And this bypass normal control of TCA cycle so NAD+ is consumed and then limit beta oxidation and ETC for to save the cell. I don’t really know, if it is so, but missing 2 of 8 (or 2 of 17 after TCA?) FADH2 seems to me not enough to be a main cause and DECR possible involvement seems to be ignored.
Miinalainen IJ, Schmitz W, Huotari A, Autio KJ, Soininen R, et al. (2009) Mitochondrial 2,4-dienoyl-CoA Reductase Deficiency in Mice Results in Severe Hypoglycemia with Stress Intolerance and Unimpaired Ketogenesis. PLoS Genet 5(7): e1000543. doi:10.1371/journal.pgen.1000543
https://pubmed.ncbi.nlm.nih.gov/19578400/
https://en.m.wikipedia.org/wiki/2,4_Dienoyl-CoA_reductase
This is an interesting comment, I had not considered this…
It looks to me like this enzyme would work in the opposite direction of superoxide. PUFA would be generating NADP+ as opposed to NADP+ being generated by SFA. I think you are underestimating the effect of the “bottleneck”. The bottleneck is not a one to one relationship, that’s why it’s so powerful. A relatively small increase in complex II input can drive a relatively large amount of superoxide.
Brad