
AKA The ROS Theory of Obesity Part 2, Part 1
Introduction
In The ROS Theory Of Obesity, I argued that the key determinant of body fatness was the saturation level of fat as it enters the mitochondria. In The Croissant Diet I addressed the dietary half of this equation, arguing that to lose weight you should avoid dietary polyunsaturated fat (PUFA) as strictly as possible, limit dietary monounsaturated fat (MUFA) and focus on consuming long chain saturated fat. I especially argued in favor of an 18 carbon length saturated fat called stearic acid which had been shown to stimulate mitochondrial fusion in humans and to eliminate abdominal fat in mice.
This article takes on the other half of the equation – your stored body fat. The fat that enters the mitochondria is a blend of recently consumed dietary fat and stored body fat. Insulin and a hormone called leptin, released by fat cells, control long term fat storage levels by manipulating the level of saturation of body fat via their effects on an enzyme called stearoyl-CoA desaturase (SCD1). SCD1 only has one function. It turns long chain saturated fats into long chain monounsaturated fats. Insulin is an up-regulator of SCD1 and leptin down-regulates it. Insulin pushes your body fat towards unsaturation which causes you to store fat. Leptin pushes your body fat towards saturation which causes you to burn fat.
In part 1 of this series I will talk about the science behind how insulin and leptin control SCD1 and how leptin prevents obesity by stimulating thermogenesis – burning off extra calories as heat. I argue that when body fat becomes too unsaturated an organism loses the ability to do leptin-stimulated thermogenesis, body temperature drops, metabolic rate drops and the organism becomes fat.
In part 2 I will argue that the difference between a lean metabolism and a post-obese metabolism is permanently up-regulated SCD1 in the post-obese state. I will present a mechanism by which dietary PUFA could lead to permanently up-regulated SCD1. I will look at dietary and supplemental factors that control SCD1 expression, how you can test to see what your levels of SCD1 are and introduce a natural inhibitor of the enzyme.
I have argued that a theory of obesity should explain a variety of diets across the human spectrum of existence and their effects on body fatness. In part 2 of this series I will additionally show that the SCD1 Theory of Obesity explains a great number of these scenarios.
Part 3 looks at how dietary monounsaturated fat (MUFA) fits in.
Basics of SCD1
One of the things I love about this system is how simple SCD1 itself is. Here is the only thing that it does: convert long chain saturated fats into long chain monounsaturated fats. It converts stearic acid – a long chain (18 carbon) saturated fat found in this such as beef suet, chocolate and my stearic acid enhanced butteroil – into oleic acid – a monounsaturated fat found in olive oil and fatty pork such as that from the Mangalitsa breed. It also converts the 16 carbon saturated fat palmitic acid into the 16 carbon monounsaturated palmitoleic acid, but at a much lower rate.
The SCD1 enzyme is also simple in its regulation. It doesn’t need to be phosphorylated to work, it’s not redox controlled. When it’s present, it just makes a double bond in long chain saturated fats. It is present whenever the RNA that codes for the gene is being made. The RNA and the protein both have short half lives – so soon after insulin stops signalling the enzyme fades away.
So all of the action is whether or not the RNA that encodes the protein is being produced (transcribed) or not. In a way this is a simple system to understand but the dynamics of SCD1 production can be quite dramatic. Just as a for instance, here is the SCD1 response in the liver to putting a fasted mouse on a fat free diet. A 14 fold increase in 24 hours!1,2
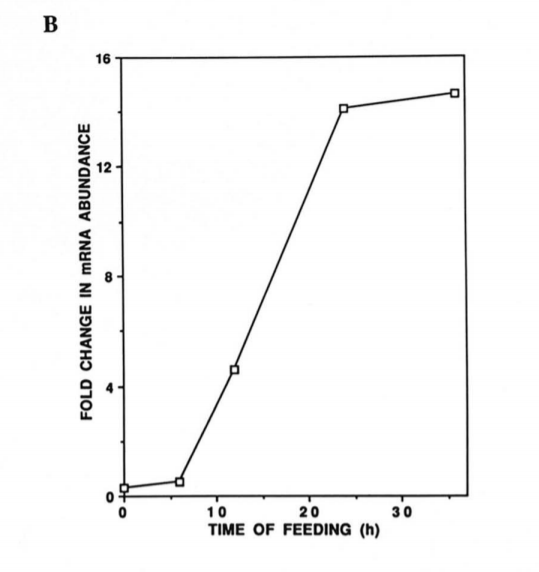
SCD1 Controls Fatness
I mentioned in my series The ROS Theory Of Obesity that obese humans produce a lot of SCD1. In fact, all kinds of things that accumulate fat have high levels of SCD1 when you look. Things that lack SCD1 typically don’t accumulate fat and simply forcing something to make SCD1 is sufficient for it to accumulate fat.
When You Look at Fat Things They Make a Lot Of SCD1
I’ve often mentioned that the Mangalitsa Breed of pig is very fatty and renowned for the fact that lard from pigs of this breed is high in monounsaturated fat. I haven’t been able to find SCD expression data for Mangalitsa but a 2008 study showed that the fatty Chinese Xiang breed of pig makes twice as much SCD1 as the much leaner large white breed.3
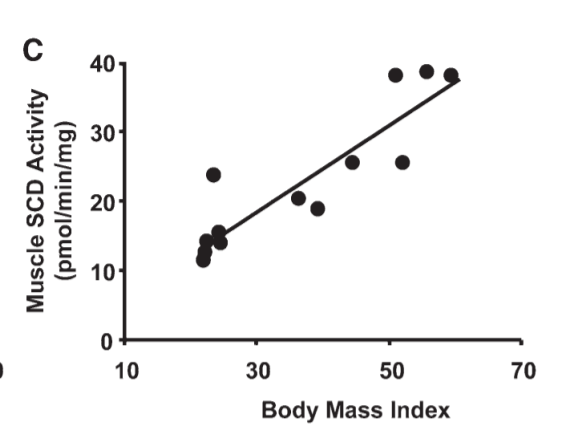
Another place we see fat accumulate is in the muscle tissue of obese humans. Guess what? Muscle tissues of obese humans make a lot more SCD1 than in lean humans.4
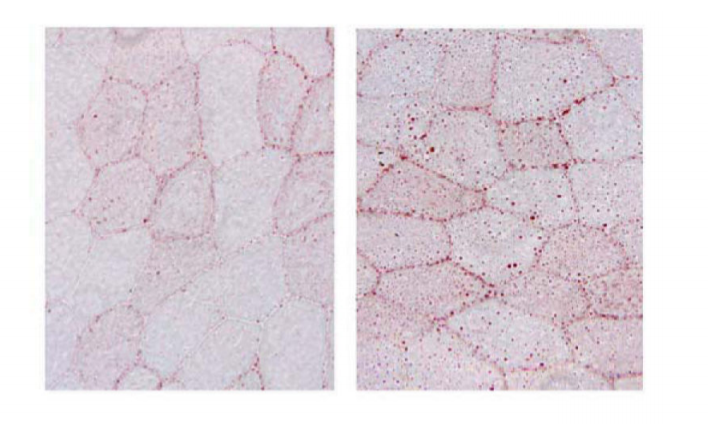
Things That Lack SCD1 Don’t Store Much Fat
I tend to think evolutionarily and I like things that have been conserved for long periods of time. Things that are conserved over time are important.
I’m going to go back to my favorite worm, the nematode C. elegans. C elegans with mutations (fat-6;fat-7) in their SCD1 gene are not able to create full lipid droplets – upper right photo. Feeding them monounsaturated fat (“oleate”, which is oleic acid such as that predominate in olive oil) restores their ability to store fat – bottom right photo.5
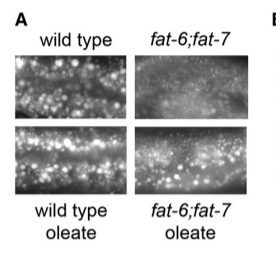
SCD1 Overexpression is Sufficient To Induce Fat Storage
Overexpressing SCD1 in cardiac myocytes – heart muscle – is sufficient for them to accumulate fat in the presence of palmitic acid: a saturated fat analogous to the Free Fatty Acids that your heart cells would be exposed to.6
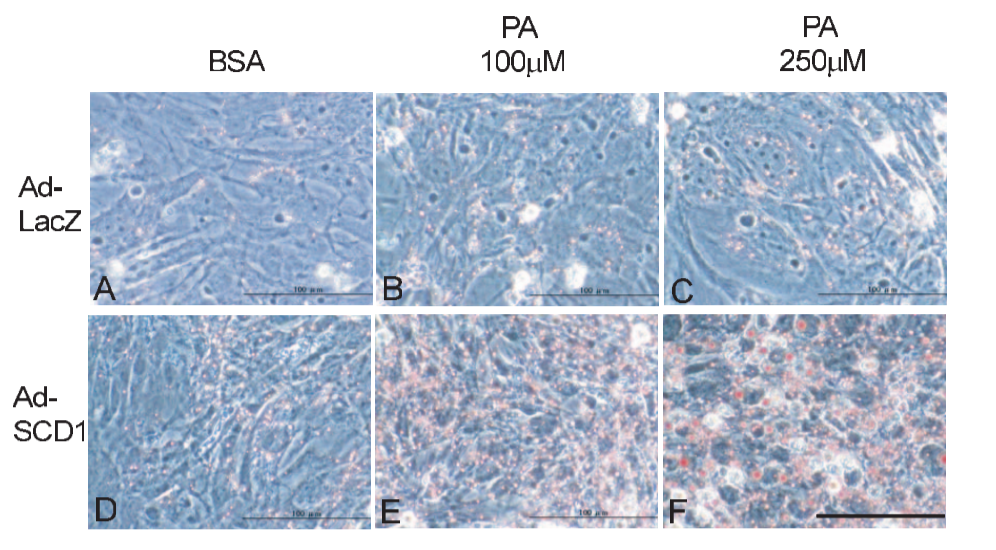
Those are heart cells. The top row makes a normal amount of SCD1 and does not accumulate fat. The bottom row makes a LOT of SCD1 and when fat is available – the amount of fat goes from 0 in the left column and increases as you move right – the cells accumulate fat. You can see the accumulated fat in E and F.
Insulin, Leptin and SCD1
It has been known since the mid-nineties that mice who are lacking the leptin gene – a hormone released by fat cells – become obese, diabetic, have a low metabolic rate and a low body temperature7. They also overeat. Leptin is a molecule that signals satiety in the hypothalamus and quickly it was shown that injecting leptin into leptin-deficient mice reduced their food intake dramatically.1 It also was shown to reduce food intake in normal mice.8
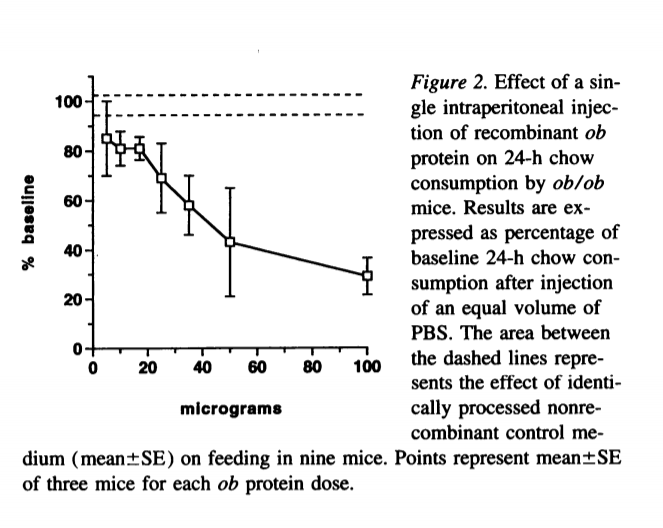
By 1998, it was shown that humans with a mutation in the leptin receptor had early onset obesity, reduced energy expenditure and low thyroid function.9 This became an incredibly hot area of obesity research, human weight-loss trials testing the effects of injected leptin were rapidly set up. And failed.10
By 2009 or so, it seems that most interest in leptin as a therapeutic tool had waned.
Most of What Leptin Does is Downregulate SCD1
When most people think of leptin, they think of apetite signalling in the hypothalamus. This dovetails well with the “calories in, calories out” school of thought about obesity. Fat people are fat because we overeat, the thinking goes. Perhaps we overeat because we are leptin resistant, perhaps because modern food is simply too palatable, perhaps it is simply because we are gluttonous. We’re probably also lazy.
Appetite regulation is only half of the equation and it seems to be the less important half. In 2002, this paper11 showed that the negative effects of leptin on body weight and metabolic rate could be mostly reversed as long as a gene called SCD1 wasn’t functional.
Look at the charts from that paper. IMPORTANT POINT: The first column is not the control mice. I find this confusing, but there you have it. The “pink” bars anre normal mice. The yellow bars (on the right) are mice lacking a functional SCD1 gene. The green bars are mice lacking both leptin AND SCD1. The blue bars have a functional SCD1 gene but lack leptin.
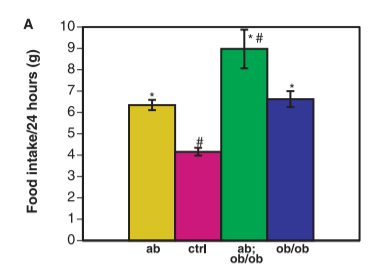
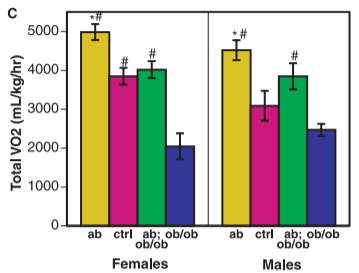
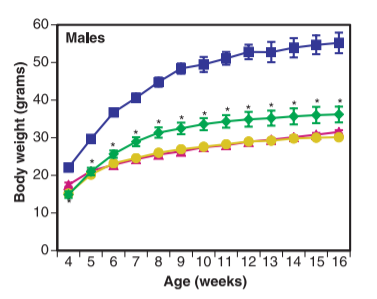
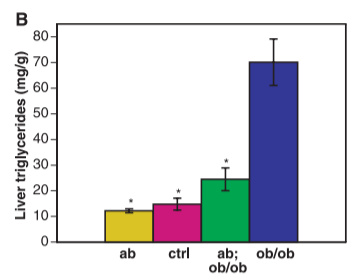
Looking at the food intake and body weight graphs, you can see – going from the pink to the blue bars – that mice lacking leptin do eat much more than normal mice and become very obese. The graph of “Total VO2” is measuring metabolic rate. You can see – going from the pink to the blue bars – that mice lacking leptin have a significantly lowered metabolic rate from normal mice. The “liver triglycerides” graph shows – going from pink to blue again – that mice lacking leptin have tremendously fatty livers compared to normal mice.
Now lets look at the yellow bars – the mice lacking SCD1 with functional leptin. They have significantly elevated metabolic rate and food intake, are as lean as control mice and have less liver fat.
Finally, let’s consider the mice lacking both SCD1 and leptin – the green bars. They indeed have little appetite control, consuming more than double the food of normal mice!! This does cause them to be a little fatter than control mice and have elevated liver fat, but not NEARLY so much as the blue bars. Their metabolism is jacked! Despite the fact that they are eating an insane amount of food, most of the extra calories are simply burned off.
Still, though, their metabolic rate isn’t as high as mice with leptin who lack SCD1.
Insulin and Leptin are in a Battle Over SCD1 Levels
Leptin directly down-regulates SCD1 in humans. It works in opposition to insulin, which up-regulates SCD1. This paper is very good.12 The authors incubated human HepG2 cells – a line of liver carcinoma cells commonly used to test metabolic effects – with either insulin, leptin or both for 24 hours. In this graph the control is on the left.
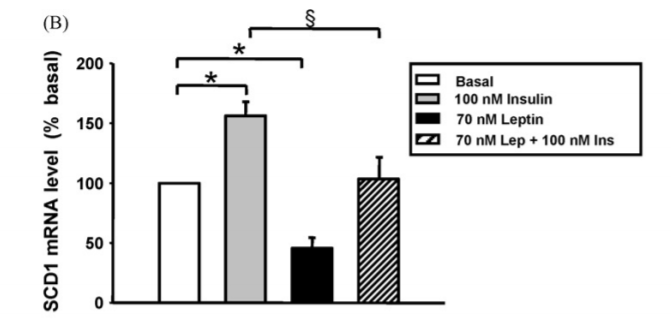
Insulin caused SCD1 to rise by 50%, leptin caused SCD1 to fall by 50% and in the presence of both the level was unchanged.
In addition to this, the authors do some very elegant biology to show that it is the ERK/MAP Kinase (MAPK) signalling pathway that is responsible for down-regulating SCD1 in response to leptin.
The Mechanism by Which Leptin Decreases SCD1 is ROS Generation in the Mitochondria
The Randle cycle controls whether mitochondria are burning fat of carbohydrates. I don’t want to go into the whole thing here, but the short version is that byproducts of glucose metabolism inhibit fat oxidation and vice versa. It seems that leptin exerts it’s effects in fat tissues by tilting the Randle cycle in favor of fat burning.13
If you’ve read The ROS Theory of Obesity you’ll know that the physiologically appropriate effect of increasing fat oxidation is mitochondrial production of ROS, and in particular hydrogen peroxide. You will also know that the more saturated the fat being burned in the mitochondria is, the more ROS it will produce. And indeed, leptin causes the production of ROS in the mitochondria.14
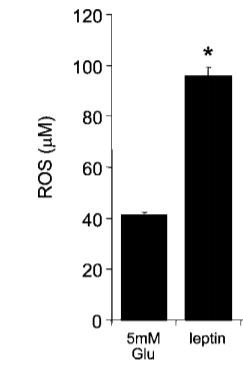
I’ve already mentioned that SCD1 levels are down-regulated by the ERK/MAPK pathway, but this seems to be an indirect type of regulation. This review about how the MAPK pathway is upregulated by ROS states quite clearly, “Oxidative stress is well known to induce the activation of ERK.” Oxidative stress is really just another term that means increased levels of ROS.
But there is a separate transcription factor that does the same thing in response to ROS production. As I talked in my post about Oxidative Balance, Nrf2 is activated in response to ROS generation. This paper built a system to study the effects of gene expression as the dose of Nrf2 steadily increased.15 It found that Nrf2 activation decreases SCD1 expression.
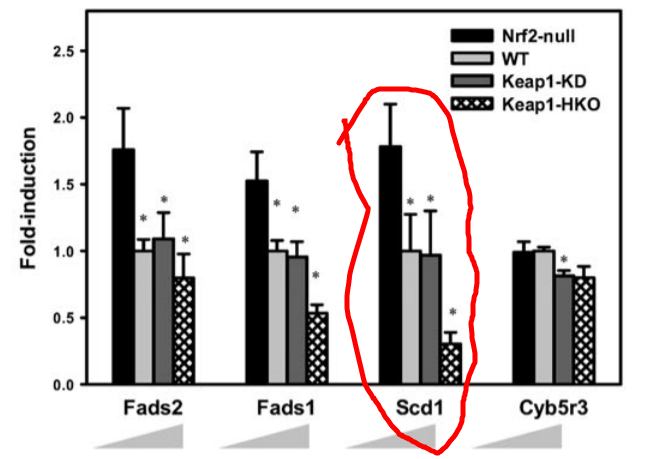
So there are two separate mechanisms in place to reduce SCD1 expression in response to a leptin induced increase in fat oxidation and the increase in ROS production it will cause.
This is A Positive Feedback Loop of ROS production
As you become fatter your body makes more leptin. The increased leptin signals to you fat cells to burn more fat. When they burn fat, it creates ROS, which stimulates the MAPK signalling pathway and Nrf2, both of which inhibit the production of SCD1. This makes your fat more saturated, which creates more ROS in the mitochondria, which stimulates the MAPK signalling pathway and Nrf2, both of which inhibit the production of SCD1. This makes your fat more saturated….
ROS Production Up-regulates UCP, Which Allows You to Burn Your Fat as Heat
I stole this diagram from here because I really like the drawing.16
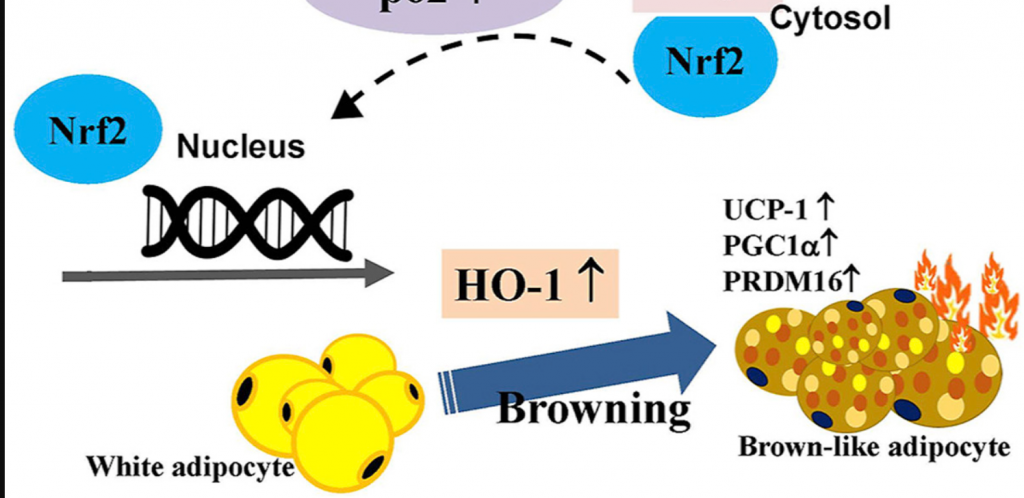
Humans have two types of adipocytes (fat cells), white and brown.17 White adipose tissue is mostly an energy storage organ. Brown adipose tissue (BAT) produces a special protein called uncoupling protein 1 (UCP-1). UCP-1 uncouples mitochondrial fat oxidation from ATP generation by allowing protons to flow back through the mitochondrial membrane.
Nrf2, which is activated in response to the positive feedback loop of ROS production, increases the expression of UCP-1 in adipose tissues, leading to the “browning” of fat.
This allows you to turn your stored fat into heat rather than stored chemical energy! You can just burn off your stored calories!
THIS is presumably the reason that SCD1 deficient mice have such higher metabolic rates than even normal mice. Their fat is highly saturated, leading to maximal ROS production, leading to Nrf2 activation, leading to UCP-1 upregulation. Then they just burn off the MASSIVE amount of calories they consume.
Sugar and SCD1
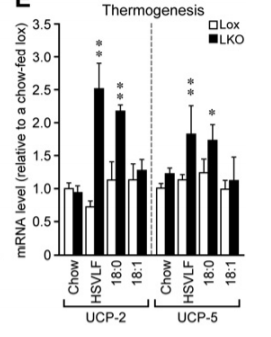
This paper has a really elegant display of this whole cycle in action.18 The authors made mice who have a liver specific deletion of the SCD1 gene. These mice are resistant to becoming obese fed a very low fat, very high sugar diet (HSVLF). (The second experiment in the paper, we’ll get to the “Western Diet” later in the series.) Fructose from the sugar is processed by the liver.
In normal mice (white bars – LKO stands for Liver KnockOut), the response to all of that sugar is to massively increase SCD1 and upregulate genes involved in de novo lipogenesis, turning that fructose into tons of oleic acid – monounsaturated fat – which is exported from the liver. They become fat on this diet.
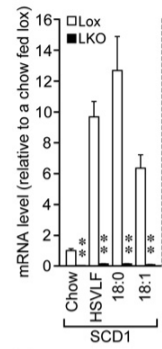
The mice lacking SCD1 (black bars) still up-regulate de novo lipogenesis in response to a high sugar diet but not nearly as much and the fat being made in and exported from the liver is of course highly saturated. These mice become very lean – leaner than the control mice. They have upregulated uncoupling proteins. They are doing thermogensis to stay lean! If you supplement their diet with the saturated fat stearic acid (18:0), they get even leaner.
But if you supplement their diet with oleic acid (18:1) – the monounsaturated product of SCD1 – they fail to upregulate uncoupling proteins and become very fat indeed!
Fat Native Americans
I’m going to end this post with an anecdote.
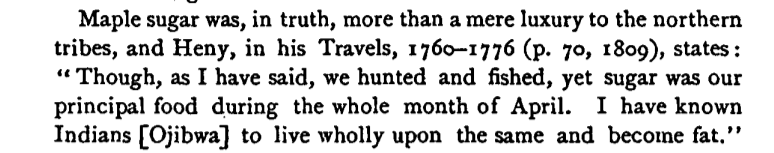
Native Americans, living an ancestral lifestyle of hunting, fishing and maple sugar making – hard work involving cutting and hauling huge quantities of firewood and buckets of sap throughout the frigid Northern winters – got fat if they ate only maple syrup.19 I think we can say this was not due to laziness. I think they become fat for the same reason the mice fed a high sugar very low fat diet did: Massive up-regulation of SCD1 and lipogenesis in their livers, which then exported huge quantities of MUFA. These native Americans would have been pathologically unable to trigger sufficient ROS production in their mitochondria when burning fat to induce UCP mediated thermogenesis.
- 1.Weigle DS, Bukowski TR, Foster DC, et al. Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J Clin Invest. Published online October 1, 1995:2065-2070. doi:10.1172/jci118254
- 2.Ntambi J. Dietary regulation of stearoyl-CoA desaturase 1 gene expression in mouse liver. J Biol Chem. 1992;267(15):10925-10930. https://www.ncbi.nlm.nih.gov/pubmed/1350282
- 3.Guo W, Wang SH, Cao HJ, et al. Gene Microarray Analysis for Porcine Adipose Tissue: Comparison of Gene Expression between Chinese Xiang Pig and Large White. Asian Australas J Anim Sci. Published online January 4, 2008:11-18. doi:10.5713/ajas.2008.60256
- 4.Hulver MW, Berggren JR, Carper MJ, et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metabolism. Published online October 2005:251-261. doi:10.1016/j.cmet.2005.09.002
- 5.Shi X, Li J, Zou X, et al. Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J Lipid Res. Published online June 20, 2013:2504-2514. doi:10.1194/jlr.m039669
- 6.Matsui H, Yokoyama T, Sekiguchi K, et al. Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS One. 2012;7(3):e33283. doi:10.1371/journal.pone.0033283
- 7.Pelleymounter M, Cullen M, Baker M, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. Published online July 28, 1995:540-543. doi:10.1126/science.7624776
- 8.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. Published online September 1, 1996:1101-1106. doi:10.1172/jci118891
- 9.Clément K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. Published online March 1998:398-401. doi:10.1038/32911
- 10.Zelissen PMJ, Stenlof K, Lean MEJ, et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: a randomized, placebo-controlled trial. Diabetes Obes Metab. Published online November 2005:755-761. doi:10.1111/j.1463-1326.2005.00468.x
- 11.Cohen P. Role for Stearoyl-CoA Desaturase-1 in Leptin-Mediated Weight Loss. Science. Published online July 12, 2002:240-243. doi:10.1126/science.1071527
- 12.Mauvoisin D, Prévost M, Ducheix S, Arnaud M-P, Mounier C. Key role of the ERK1/2 MAPK pathway in the transcriptional regulation of the Stearoyl-CoA Desaturase (SCD1) gene expression in response to leptin. Molecular and Cellular Endocrinology. Published online May 5, 2010:116-128. doi:10.1016/j.mce.2010.01.027
- 13.Wein S, Ukropec J, Gašperíková D, Klimeš I, Šeböková E. Concerted Action of Leptin in Regulation of Fatty Acid Oxidation in Skeletal Muscle and Liver. Exp Clin Endocrinol Diabetes. Published online May 3, 2007:244-251. doi:10.1055/s-2007-956166
- 14.Yamagishi S, Edelstein D, Du X, Kaneda Y, Guzmán M, Brownlee M. Leptin Induces Mitochondrial Superoxide Production and Monocyte Chemoattractant Protein-1 Expression in Aortic Endothelial Cells by Increasing Fatty Acid Oxidation via Protein Kinase A. J Biol Chem. Published online May 7, 2001:25096-25100. doi:10.1074/jbc.m007383200
- 15.Wu KC, Cui JY, Klaassen CD. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicological Sciences. Published online July 20, 2011:590-600. doi:10.1093/toxsci/kfr183
- 16.Tsai Y-C, Wang C-W, Wen B-Y, et al. Involvement of the p62/Nrf2/HO-1 pathway in the browning effect of irisin in 3T3-L1 adipocytes. Molecular and Cellular Endocrinology. Published online August 2020:110915. doi:10.1016/j.mce.2020.110915
- 17.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. American Journal of Physiology-Endocrinology and Metabolism. Published online August 2007:E444-E452. doi:10.1152/ajpendo.00691.2006
- 18.Miyazaki M, Flowers MT, Sampath H, et al. Hepatic Stearoyl-CoA Desaturase-1 Deficiency Protects Mice from Carbohydrate-Induced Adiposity and Hepatic Steatosis. Cell Metabolism. Published online December 2007:484-496. doi:10.1016/j.cmet.2007.10.014
- 19.Henshaw H. Indian Origin Of Maple Sugar. Indian Origin of Maple Sugar. Published 1890. https://anthrosource.onlinelibrary.wiley.com/doi/pdf/10.1525/aa.1890.3.4.02a00050
how to boost leptin levels? increase saturated fats? I seem to have better cold resistance after eating croissants but this may just be in my mind. Is there a way to test leptin levels? how about this test? https://www.lifeextension.com/lab-testing/itemlc146712/leptin-blood-test
I think most of us – especially if you’re overweight – have tons of leptin. Lack of leptin isn’t the issue. The issue is lack of ability to drive ROS production in the mitochondria due to insufficient fat saturation levels. Weight loss trials have shown convincingly that injecting extra leptin does nothing for weight loss if you’re already overweight.
It would be interesting for many to test this and see what the numbers look like. There is a Reddit sub (SaturatedFat) where we could post results (for easy searching later on). I am considering doing this.
Yes!
Somebody posted results this morning. I have on my list: make citizen science database of results. And everyone should check out the Reddit:
https://reddit.com/r/SaturatedFat
Just wanted to stop by and say that I am so darn thrilled I heard the podcast with Dr Paul Saladino on TCD.
In one Of those people who had a fasting insulin of 10+ after over a year of carnivore/keto eating with little to no weight loss.
I’m having a blast eating traditional foods like pasta with homemade Alfredo, oatmeal with lots of butter, cocoa butter in my decaf every morning, etc and I literally feel like a furnace for hours after eating!
I’m keeping track of my measurements and my waist is already shrinking after only a week!
This stuff is incredibly interesting to me, and I hope to be a success story so I can share this info with all those I care about!
Thank you Thank you Thank you, Brad!!!!
Thank YOU, Kelly!
Let us know how it goes!
Hi Kelly, I’ve also been adding cocoa butter to my morning decaf and blending it in. I really enjoy it. Even just added some cb to hot water with a little salt. I’m curious to know how much cb you add in to your decaf.
So how do I get my SCD1 levels lower?
Check out part 2!
https://fireinabottle.net/the-scd1-theory-of-obesity-part-2-the-post-obese-metabolism-and-what-you-can-do-about-it/
Hi Brad,
Thank you for this in-depth presentation. It makes me very confident in the theory. I read the entire post and plan to read it again. Could you break it down for us a little more, maybe a short paragraph with a conclusion and concrete steps we can take to deregulate SDC1? Or is it as simple as eat more saturated fat? If you could have the perfect eating day what would you eat to optimize the downregulation of SDC1? Maybe I don’t need to understand the biochemistry of it completely and just need to do?
Thank you in advance. I love your work and can’t wait for the low pufa pork!
Hi! Did you see part 2?
https://fireinabottle.net/the-scd1-theory-of-obesity-part-2-the-post-obese-metabolism-and-what-you-can-do-about-it/
Brad
Funny, I have a theory of obesity as well but looked at from a complete different angle. Perhaps you want to look at it and see how compatible it is with your theory.
https://designedbynature.design.blog/2020/05/13/hyprocico-the-theory-behind-obesity/
Can I pour stearic acid powder in hot coffee? Will it dissolve and blend well by just steering with spoon without a blender?
It will probably not melt unless your coffee is VERY hot. If it DOES melt, it will solidify in your mouth/as your coffee cools.
Brad
The yellow bars are on the left (not right) 😊
I struggle with left and right.
If you look at the back of your left hand with all fingers spread out the index finger and thumb will look like a capital L, like L for left. The right hand won’t. If you are dyslexic it is the side from which another letter would flow and attach easily at the base of the L… Little trick that you could do to check even in public and no one would notice.
Wow. Thanks for laying all this out. This info definitely presents a conundrum. One wonders now if saturated is what we want almost exclusively, and so olive oil and avocados, for example, truly are to be avoided? Yet the info I have says, especially with my APOE4 gene, that I need to balance saturated fats with Mufas to reduce my chances of cardiovascular and dementia.
https://pubmed.ncbi.nlm.nih.gov/8857921/
Now, the obvious detail is that this article is simply looking at obesity, not with overall system health. But is it one or the other? Are the suggestions for Mufas as safe fats misguided? Are the studies that show APOE4 being amplified by high saturated fat ratios inaccurate? I wouldn’t be surprised. Certainly we have been learning that the complete demonization of saturated fat was mostly or completely misguided. The ROS theory certainly turns more things on their heads and gets us thinking.
After reading up here, I’m now quite skeptical of the “antioxident = more better always” mentality. My vitamin C supplement intake is now down to a couple hundred grams daily, no more. But which fats to favor (if not obese) is more perplexing. Especially with an APOE4 allele. Should I favor our homeade ghee mostly or continue to use the olive/avo oil when appropriate? Hmmmm…
Oops, meant to link this one: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6213759/
Not that it’s THE study or the one that advises on diet for APOE4 folks, just a quick reference I grabbed to show that point of “view”.
That study still assumes that high cholesterol is the main culprit in CVD. I would say this has been debunked thoroughly over the years. “APOE4 genotype (carriage of ε4 alleles) has also been linked to insulin resistance and the metabolic syndrome” – from the study link. When you look at the effects metabolic syndrome has on the body over time, you have one of the major causes of Alzheimers and CVD. That diet used in the study was also high in carbs and not what I would consider a high fat diet: “The target intake for total fat was 38% of energy (%E) in the high-SF and both high-MUFA diets and 28%E in the 2 LF diets, which also had a higher target CHO intake (55 vs. 45%E).” Protein was low with only 16% on average. This is not a diet that would be helpful for someone prone to metabolic syndrome. This is the diet study that the study in the link is based on https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3594744/
From glancing at these studies I would say the contributing factor to the increased risk for coronary disease, Alzheimers in the presence of this gene is elevated insulin aka metabolic syndrome with all the complications that situation creates. When you address that, you will mitigate the risk that may come with your gene. I say may, because all of the genetic studies are still in their infancy and there is still too much guesswork. Although maintaining good metabolic function, with normal insulin levels as its base, is a good idea in general.
Again, from the study you posted: ” There is evidence to suggest that APOE4 genotype confers increased sensitivity to changes in dietary total fat, cholesterol and fatty acid composition [14,15,16,17,18,19,20], though this evidence has been confounded by underpowered studies, variation in study design and dietary intake [21]. Attempts to personalise dietary advice on the basis of APOE genotype have also met with mixed results [22,23].” The key word here is suggest. That is not hard scientific fact. The phrase ‘evidence to suggest’ couldn’t be more contradictory in and by itself.
OH GOOD GOD YOU’RE TAKING SEVERAL HUNDRED GRAMS OF VITAMIN C PER DAY?!?!
Haha. I’m kidding, I assume you mean mg.
Anyhoo: As to DIETARY MUFA, as opposed to internally produced MUFA, I have a post coming out this week explaining how it’s different and probably OK within limits.
Brad
This theory is very interesting but something does not work. SCD1 is an enzyme working like most other enzymes i.e. increased product leads to decreased activity. This means that supplying it with more oleic acid should decrease its activity. Something else is explaining the difference in weight between stearic acid fed and oleic acid animals. This probably has to do with the mitochondrial processing.
Another problem in general is when animals are fed 50% of calories in PUFAs, 0% carb and the rest protein they dont get fat at all. Possibly this is due to “rabbit starvation”.
SCD1 seems to be down-regulated by one of its products – palmitoleic acid – which is fairly rare from dietary sources but not by oleic acid which is very common from dietary sources. This is sensible, I think.
Getting 50% of calories from PUFA wuld likely cause massive oxidative stress due to generation from, for instance, 4-HNE. This would be equivalent to massive ROS generation but it’s not a road I’d recommend going down.
Brad
Thanks for a great post. I’m surprised to learn what I believe you are describing as a negative for Mangalitsa pork. I know it to be one of the favorite fatty meat recommendations at the Paleo Medicina clinic in Hungary. This well known group uses a very high fat dietary approach (termed PKD – Paleolithic Ketogenic Diet), 2:1 in grams of fat to protein for the management of many chronic and autoimmune problems. Dr. Zofia Clemens has presented often about their approach.
There’s a different between dietary MUFA and MUFA that you make in your body. Post coming next week. The mangalitsa pigs are fat because they make MUFA in their bodies.
Brad
I purchased the Butteroil product about a month ago. I had issues with incorporating it into carb food because of my perception of the ‘taste’. I decide instead to form 1/4 tsp size portions, refrigerate them, then swallow multiple portions (maybe a couple of tsp worth) throughout the day like a supplement. A few of my supplements require fat to metabolize so I figured … what the heck..I love a good N=1. OMG, the impact is amazing. It has dramatically killed my appetite, waist circumference is down and of course I have lost weight. I have eaten a bit of carbs with my meals (Delicata squash, sour dough bread) but again once I have a small portion .. I have no desire for more. I have about 20 more pounds to lose so I excited to continue. Thank you, Brad and this group for all you do to support metabolic health!
Hi Mary!
TI’m so glad it’s helping for you! The good news is I’m working on a more natural highly saturated butter product that will similar to the current one but with a much more natural butter flavor! I’m hoping to have it available in the first half of 2021!
Brad
Brilliant idea! How much do you take in a day?
So SCD1 affects type of FA made from starch 18:0 or 18:1
But does it affect dietary SFA? will it turn SA from my cocoa butter into 18:1?
Brad covers it in part 2. He said this about himself: “Clearly my fat cells have been busy converting all of that stearic acid I’ve been eating into oleic acid. I am trapped on the wrong side of the mountain!”