
Summary
- Activating AMPK in the livers of mice prevents diet induced obesity despite higher caloric consumption
- Mice with activated AMPK have lower levels of cytochrome P450 enzymes that produce 12-HETE and 15-HETE – specific activators of PPAR gamma
- Mice with activated AMPK have higher rates of both fat and glucose metabolism
- AMPK blocks the pathway from dietary PUFA to the torpid metabolism at nearly every step
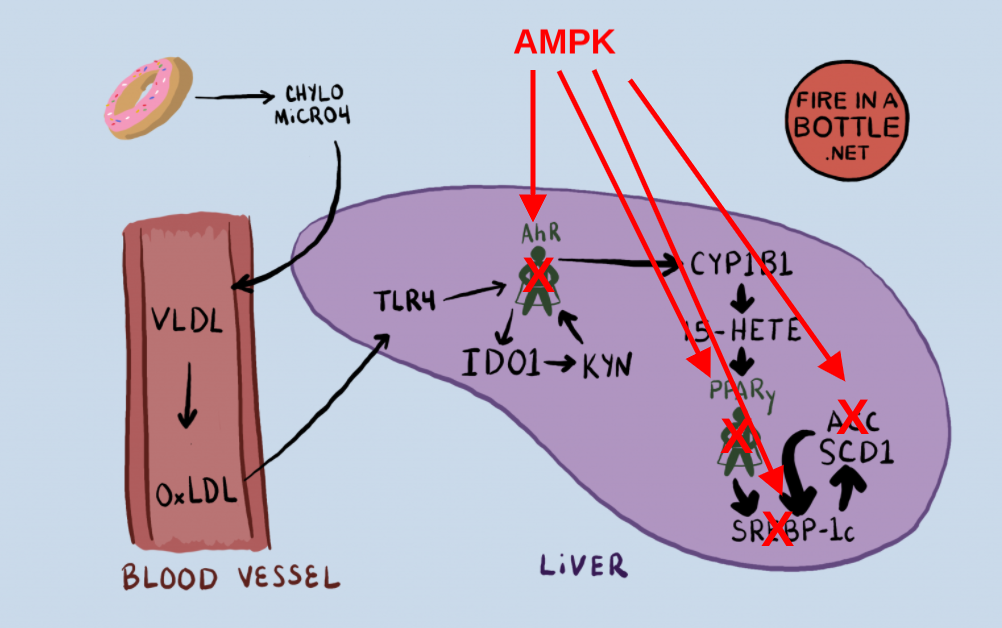
In the last post I laid out the mechanism by which I believe the lipogenic metabolism is triggered. In short, Dietary PUFA creates oxidized LDL which triggers the Aryl Hydrocarbon receptor which creates ligands such as 12-HETE and 15-HETE which induce PPAR gamma activation in the liver.
In this 2019 paper1, the authors created a mouse model where they could turn on high AMPK activity in the liver of mice in response to feeding the mice the antibiotic doxycycline. It’s a clever system. The mice who have highly activated AMPK in their liver are resistant to diet induced obesity.
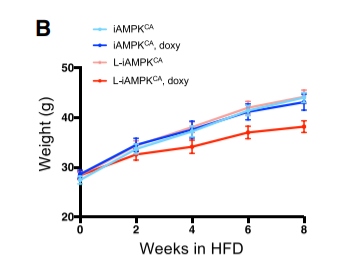
The authors looked at the genes which were the most DOWN-regulated (turned off) in response to AMPK activation. You have to dig into the supplemental figures to find out, but amongst the genes that AMPK down-regulated the most are the cytochrome P450 enzymes Cyp4a14, Cyp2c37, Cyp2c50 and Cyp1a2. Guess what all of these enzymes do? They make oxidized PUFA, such as 12-HETE, 15-HETE and 20-HETE, which are known activators of PPAR gamma.2,3
Activated AMPK suppresses the upregulation of cytochrome P450 enzymes which provide the key to a torpid metabolism.
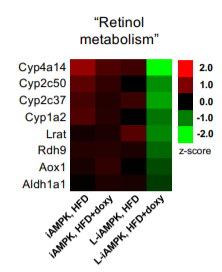
Eat More, Weigh Less
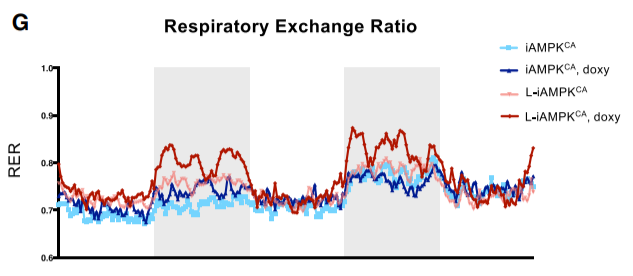
Also buried in the supplemental section is a graph showing that the lean mice with activated AMPK ate the most food and therefore the most calories. So the gluttonous mice were the leanest because the root cause of obesity is not calories-in-calories-out, it’s enzyme systems.
The lean mice with activated AMPK had the highest rates of fat oxidation, but they ALSO had the highest RER during the nighttime when mice are active. This shows that they made better use of glucose as a fuel. They were better fat burners AND they were better carbohydrate burners. They had metabolic flexibility, the same thing which is lost in obese humans.
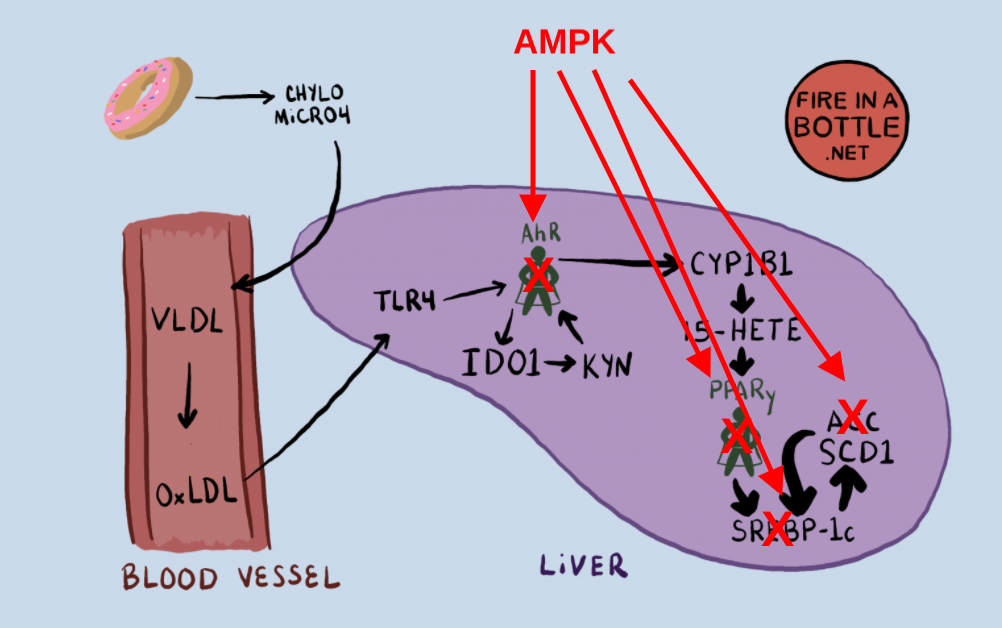
How does AMPK do this?
AMPK blocks my proposed pathway from PUFA consumption to a torpid metabolism in at least four spots. The first two are straight forward. AMPK phosphorylates the enzymes ACC4 and SREBP-1c5 and deactivates them.
SREBP-1c is the transcription factor that turns on the lipogenic genes ACC, FASN, SCD1 and ELOVL6, all of which combine to create oleic acid which upregulates SREBP-1c. By inactivating SREBP-1c, AMPK breaks this positive feedback loop.
ACC is responsible for converting acetyl-CoA – the product of the breakdown of glucose and fat – into malonyl-CoA, the starting point of lipogenesis. Malonyl-CoA also supresses the activity of CPT16 – the enzyme responsible for shuttling fat into the mitochondria – and therefore slows down fat burning.
The Nuclear Receptor Superfamily
The third mechanism by which AMPK blocks the torpid metabolism is more complicated. PPAR gamma and AhR are both members of the nuclear receptor superfamily. The nuclear receptors are transcription factors found in the cytosol (cell water). They recognize different “ligands”. Ligands are molecules that bind to the nuclear receptors.
Ligands that turn nuclear receptors OFF are called antagonists and ligands that turn them on are called agonists.
12-HETE is a PPAR gamma agonist – a ligand that turns it on. When PPAR gamma binds 12-HETE it moves into the nucleus. THEN it forms a dimer with something called RXR – the retinoid X receptor – which is another member of the nuclear receptor superfamily that is activated by vitamin A. A dimer means that the two nuclear receptors come together like two halves of a walnut shell. When BOTH RXR and PPAR gamma are activated they form a dimer and they turn genes ON.
The Obesogen Tributyltin
I’m using the example of RXR and PPAR gamma because within the last 20 years the term “obesogen” was created. The first recognized obesogen was something called Tributyltin. Tributyltin was used for decades to coat the bottom of boats at sea to prevent things from growing on the hulls of the boats. This practice led to the disturbance of many marine ecosystems.
Perhaps you’ve already guessed, but tributyltin is a dual agonist of both PPAR gamma and RXR.7 If you activate PPAR gamma and it’s dimer partner RXR, it makes things fat. If you are a strict interpreter of the calories-in-calories-out school of obesity, you then must conclude that co-activation of PPAR gamma and RXR makes animals slothful and gluttonous.
Shutting Down The Nuclear Receptors
Many nuclear receptors form a dimer with RXR, but the AhR has it’s own dimer partner called ARNT.8 When the AhR is activated it binds ARNT and they go together and turn on the cytochrome P450 enzyme which oxidize the PUFA that becomes the agonist of PPAR gamma.
AMPK up-regulates a protein called Small Heterodimer Partner (SHP)9. SHP is a member of the nuclear receptor superfamily which doesn’t have a DNA binding domain and can’t turn genes on. SHP can bind to both RXR and ARNT. When it binds these, they are inactivated.
AMPK activates a protein that inactivates the binding partners of both PPAR gamma and the AhR. SHP is in fact a sort of global repressor of nuclear receptor superfamily activity, also binding the liver X receptor, the dimer partner of SREBP-1c, which is also a member of the nuclear receptor superfamily. AMPK is doubly deactivating SREBP-1c, by phosphorylating it and by increasing SHP to compete for its binding partner.
Summary
OK. That got a little heavy on the molecular biology. The good news is that you don’t have to memorize all of this. Here’s the take home:
Dietary PUFA sets off a chain of events leading to activation of the AhR which provides the oxidized PUFA necessary to become torpid and increase the expression of lipogenic genes. Activated AMPK can block this pathway at nearly every step. Which is to say that activated AMPK is the kryptonite of the nuclear receptor superfamily, who are responsible for turning on torpor. That is why the mice with activated AMPK and resistance to obesity had low levels of cytochrome P450 enzymes in their livers.
We’ve already talked on this blog about a couple ways to activate AMPK, including the diabetes drug metformin and the Chinese herb berberine. The next couple articles I am going to dive into other causes of AMPK up or down-regulation.
- 1.Garcia D, Hellberg K, Chaix A, et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Reports. Published online January 2019:192-208.e6. doi:10.1016/j.celrep.2018.12.036
- 2.Luo G, Zeldin DC, Blaisdell JA, Hodgson E, Goldstein JA. Cloning and Expression of Murine CYP2Cs and Their Ability to Metabolize Arachidonic Acid. Archives of Biochemistry and Biophysics. Published online September 1998:45-57. doi:10.1006/abbi.1998.0806
- 3.Gilani A, Pandey V, Garcia V, et al. High-fat diet-induced obesity and insulin resistance in CYP4a14−/− mice is mediated by 20-HETE. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. Published online November 1, 2018:R934-R944. doi:10.1152/ajpregu.00125.2018
- 4.Peterson JM, Aja S, Wei Z, Wong GW. CTRP1 Protein Enhances Fatty Acid Oxidation via AMP-activated Protein Kinase (AMPK) Activation and Acetyl-CoA Carboxylase (ACC) Inhibition. Journal of Biological Chemistry. Published online January 2012:1576-1587. doi:10.1074/jbc.m111.278333
- 5.Li Y, Xu S, Mihaylova MM, et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metabolism. Published online April 2011:376-388. doi:10.1016/j.cmet.2011.03.009
- 6.López-Viñas E, Bentebibel A, Gurunathan C, et al. Definition by Functional and Structural Analysis of Two Malonyl-CoA Sites in Carnitine Palmitoyltransferase 1A. Journal of Biological Chemistry. Published online June 2007:18212-18224. doi:10.1074/jbc.m700885200
- 7.Grün F. The Obesogen Tributyltin. In: Vitamins & Hormones. Elsevier; 2014:277-325. doi:10.1016/b978-0-12-800095-3.00011-0
- 8.Swanson HI. DNA binding and protein interactions of the AHR/ARNT heterodimer that facilitate gene activation. Chemico-Biological Interactions. Published online September 2002:63-76. doi:10.1016/s0009-2797(02)00066-2
- 9.Lee J-M, Seo W-Y, Song K-H, et al. AMPK-dependent Repression of Hepatic Gluconeogenesis via Disruption of CREB·CRTC2 Complex by Orphan Nuclear Receptor Small Heterodimer Partner. Journal of Biological Chemistry. Published online October 2010:32182-32191. doi:10.1074/jbc.m110.134890
A lot of Ray Peat is in this analysis. If you become carb intolerant you are going dowwnnnn!
I’m not a strict CICO person per se but wouldn’t all of this still fall under that category? A lot of this is speaking about down regulation of metabolism so wouldn’t that be lowering CO and correcting this increasing CO? I think what most of the CICO people struggle with is they pay too much attention to the CI side of the equation.
That is one way to look at it, but there is also a physical reality. Fat needs to be moved from fat cells to the mitochondria and burned to lose weight. If you don’t have the enzymes in place to do this, you will stay fat unless you literally starve yourself. You can call getting your enzymes right “increasing metabolic rate”, but if your liver is making a lot of oleic acid you’re going to have fatty liver and CICO will have no effect on it. It’s a physical reality.
Brad thank you for all you do!
You’re welcome!
So I guess the bottom line is to eat sufficient protein, and starches (not more per gram than the fat) loaded with saturated fat?
Certainly that is the concept of The Croissant Diet. And it’s the basis of the traditional French and Amercian diets. It won’t necessarily work if your SCD1 is upregulated.
Brad, you’re on a streak!
“Activating AMPK in the livers of mice prevents diet induced obesity despite higher caloric consumption”.
Induced or prevented?
If ever there was an explanation for Warren Buffet’s wisdom- If you want to feel like a kid, eat like one.
Kraft Mac & Cheese in particular uses turmeric for the color of the cheese “sauce”.
https://pubmed.ncbi.nlm.nih.gov/19665995/
The ole staple of kid dinners has an AMPK agonist in it.
If Curcumin is also an anti-oxidant, doesn’t that interfere with ROS signalling – which would be a bad thing in the context of the croissant diet?
Most things that are “antioxidants” actually produce ROS and then the body responds my generating uncoupling proteins, which help to increase metabolic rate. So when your hear “antioxidant” replace it with the phrase “reodx active”.
Ok so since Brad wrote this blog post and shined the light on AMPK, I’ve experimented with a few from what I’ve researched – and suggested to me by others…
Hibiscus tea: I drink 4 teabags’ worth (steeped in 32oz water overnight in the fridge) every day, especially before meals, and it’s made one of the biggest transformations I’ve seen yet. Started about 1 week ago.
Article: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5579700/
Turmeric: Curcumin is apparently an AMPK agonist as well. https://www.karger.com/Article/Fulltext/354516
Past 2 days I’ve done a morning routine that is absolutely CRAZY, even manic:
9/7: Omelette made with 3 eggs, goat cheese, preserved lemons, onion, about 1/2tsp turmeric, cooked in stearic acid:butter (~1:1). Drank 1 cup hibiscus tea, some coffee with the omelette. Sterculia oil + fish oil + astaxanthin taken immediately before.
By the time I got to the freeway on the drive in to work, I was almost overloaded with energy I felt slight butterflies in my stomach and slightly dizzy when I got to work, was overwhelming. Fidgeting the whole way.
9/8 (today): 1/3lb grass fed burger cooked on cast iron, 1 slice of cheese, an egg fried in the burger grease. 1 cup hibiscus tea before even starting to prepare the burger, coffee with the burger, and an orange afterwards. Sterculia oil + fish oil + astaxanthin taken immediately after. Drinking a mix of 1/2c hibiscus tea and water (in my water bottle) afterwards.
Currently sitting at my desk with my feet up on their tippy-toes dancing with a feeling of overwhelming energy. Heart rate 83, not super high but definitely not “relaxed” sitting at my desk…
I’ve tried most of these things before – the burger+cheese+egg is awesome, and I’ve had coffee in recent weeks, the sterculia+fish oil+astaxanthin & frequent consumption of oranges throughout the day is something I’ve done for months. I’ve never seen this level of “omg” tripping balls energy after eating this combination – Stearic acid might have got me close before but usually it’s a feeling of rock-hard satiety. The real energy surge from stearic acid I’ve seen was making biscuits with it – huge mix of starch + stearic acid seems to help if I haven’t done much carbs lately. I was doing turmeric golden milk with an artificial sweetener for a couple weeks, was a thing, didn’t notice any huge rush of energy.
The hibiscus tea is the big change this past week and it’s made a DRAMATIC effect on my energy levels. I notice a similar albeit muted version of this after lunch – instead of feeling “tired” after eating I’m revved up and have to take a long walk. I haven’t had a stearic acid-rich lunch yet though. Dinner similar deal, if I drink hibiscus tea too late I end up having to take another herb (like Jiaogulan) to help me get to sleep.
That’s amazing, it’s been a couple weeks now, are you still seeing the same thing with the Hibiscus tea?
The hibiscus is still having a thermogenic effect for sure. Seems I get fidgety a lot after eating. Weight loss isn’t necessarily happening, seems I dropped 2lb and stalled.
I’ve settled into a routine: Breakfast cheeseburger every morning with hibiscus tea, coffee, and an orange. Power breakfast if I’ve ever seen one!
I have been consuming sugar a bit more in the past 2wk so I suspect that is preventing weight loss. I believe I am a hyper-SCD1 upregulator in response to sugar.
I find the theories here intriguing but I am also researching the Testosterone role.
I came across this paper:
https://joe.bioscientifica.com/view/journals/joe/217/3/R25.xml?body=contentSummary-10171
A grand unifying theory of obesity it seems to me must include discussion of T. Being fat lowers your T. Lower T makes it easier to get fatter.
Male T levels have fallen by nearly 50% over the last 50 years population wide. Pretty important to account for. Not going to solve the population wide issue without it.
I’m sure that plays a role. I also suspect (having done literally zero research on it) that testosterone’s role in this will be sub-survient to ROS/PPARy/torpor, etc. We shall see!
Brad