
Obesogens are environmental toxins that make you fat. A commonality among them is that they activate receptors such as hormone receptors, androgen receptors, and perhaps most importantly, receptors in the nuclear receptor superfamily. Of course, if certain chemicals can make you fat, you have to really question the calories-in-calories-out theory of obesity. Certain chemicals can make you fat.
Classic examples of obesogens1–5:
The nuclear receptor superfamily members have several things in common6–12:
Omega-6 and omega-3 oxylipins are implicated in soybean oil-induced obesity in mice
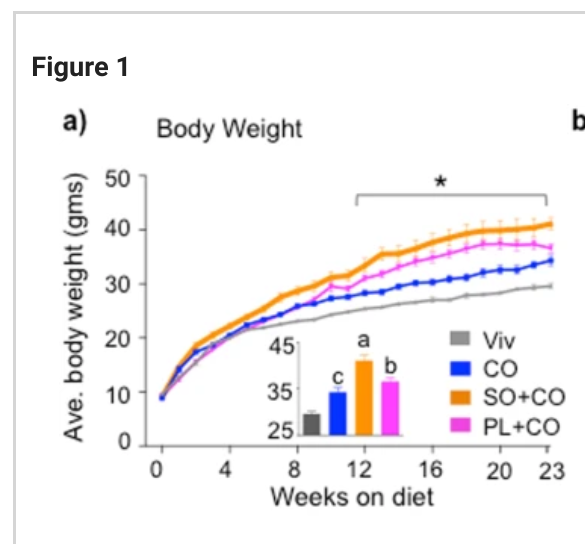
Let’s consider the findings of this paper keeping in mind the idea of PUFA as obesogens that stimulate cytochrome p450s, create oxylipins and reduce NAD+. Here are the bodyweights of mice fed a lowfat diet (Viv – grey), a diet with hydrogenated coconut oil (CO – blue), a diet with more MUFA (PL+CO – pink) and a diet with more PUFA as a 50:50 mix of coconut oil with soybean oil (SO+CO – orange). As you can see, the coconut oil fed mice are the leanest ones who were given a high fat diet and the soybean oil fed ones are the fattest.
There are significant effects of soybean oil compared to coconut oil on activation of nuclear receptor family members as we can see by the changes in regulation of cytochrome p450 enzymes. Cyp3a41a is upregulated. The Cyp3a (cytochrome P450 3a) family is primarily controlled by the SXR13. We also see downregulation of Cyp4a12a. The Cyp4a family is primarily controlled by PPAR alpha. This pattern suggests that soybean oil is activating the SXR and de-activating PPAR alpha (even though it is a known PPAR alpha activator, it’s complicated).
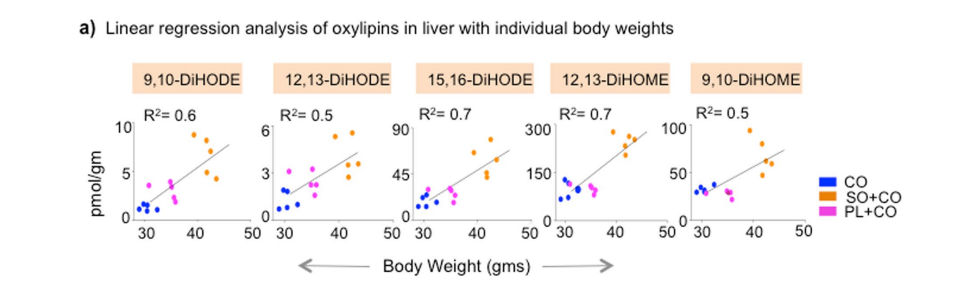
The authors of the paper spend a lot of time talking about the relationship between the weight gain in soybean oil fed animals with the oxylipins 9,10 EpODE, 15,16 EpODE, 9,10 EpOME and 12,13 EpOME. Which is fair, these epoxides were all upregulated by soybean oil, correlate with levels of obesity and are known or probable PPAR gamma activators.
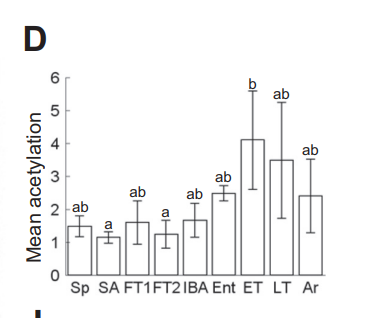
Still, my eye couldn’t help but be drawn to the upregulation of 12-HETE by soybean oil in this “MAP” of the findings. Green dots mean “higher in animals fed soybean oil compared to those fed MUFA”.
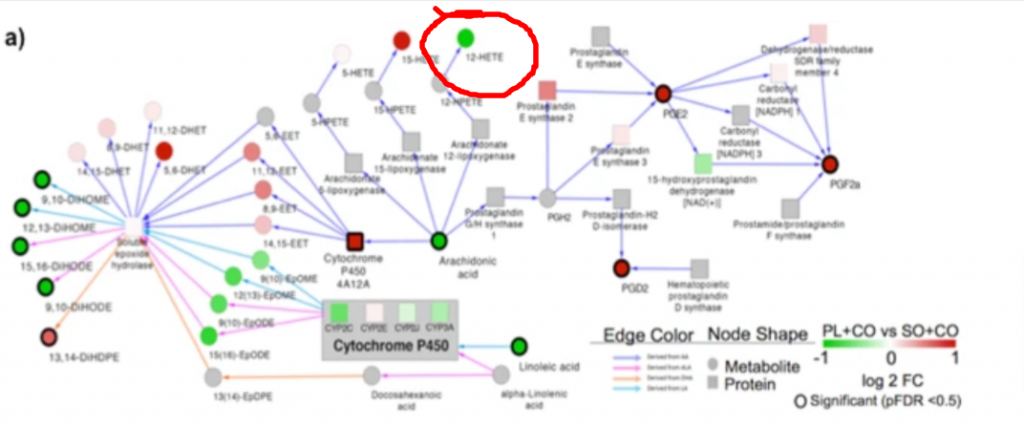
12-HETE is a known strong activator of BOTH PPAR gamma8 and the AhR14. PPAR gamma upregulates SCD115 and the NAD+ dependent enzyme CD3811. The AhR upregulates SCD116 and the NAD+ dependent enzyme PARP17.
SCD1, CD38 and PARP all decrease available levels of NAD+, bringing the metabolic rate to a crawl and increasing acetyl-CoA levels once again. As I’ve already mentioned, PPAR gamma activators and AhR activators are both known obesogens, as are those of the SXR. What better way to encourage fat storage than to upregulate lipogenic enzymes while at the same time depressing NAD+ levels?
We can say that PUFA is acting like an (is an?) obesogen by stimulating multiple nuclear receptors. The mice given the soybean oil DID get the fattest….
Torpor Time
None of this is pathology. This is actually a beautiful example of biological regulation. Consider once again the hibernating animal. Its biological imperative is to store fat for winter once the signal comes. The signal is the ripening of acorns, which introduce significant MUFA and PUFA into the diet. The response to the unsaturated fats is multi-layered and self-reinforcing, ensuring the body stores sufficient fat.
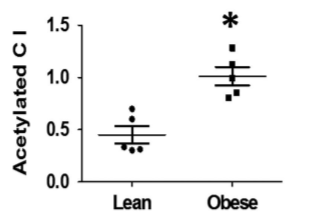
Level 1: The MUFA and PUFA in the acorns cause a slowdown in the mitochondria by flooding the mitochondria with fat and reducing production of NAD+ via the superoxide/glutathione reductase/NNT pathway. The slowdown in metabolic rate leads to a buildup of acetyl-CoA, which activates lipogenic enzymes PPAR gamma and SREBP-1c and deactivates mitochondrial enzymes involved in metabolism, adding to the slowdown. All of this is a direct mitochondrial effect of unsaturated fats.
Level 2: The PUFA in the acorns act as obesogens, having a dramatic effect on nuclear receptor signaling. It is unclear what the exact chain of events is, but the SXR is activated as are presumably PPAR gamma and the AhR, at least. PPAR gamma and the AhR upregulate CD38 and PARP, respectively, lowering NAD+ levels even further. Low NAD+ levels lead to rising acetyl-CoA levels and increasing acetylation of both lipogenic transcription factors (activating them) and mitochondrial enzymes (deactivating them). You see how the unburnt fuel is the very thing that triggers fat storage rather than fat burning?
Level 3: The steadily increasing activation level of lipogenic enzymes lead to a progressive rise in SCD1 levels. SCD1 is upregulated by both PPAR gamma and SREBP-1c, which are directly activated by rising acetyl-CoA levels. SCD1 unsaturates your body fat and when it’s fully activated it limits your ability to generate NAD+ through the superoxide/Glutathione Reductase/NNT pathway, leading to increased acetyl-CoA levels. Hibernating animals and obese humans are full of MUFA from SCD1.
The end result of this is a progressive increase in SCD1 levels and mitochondrial acetylation levels as obesity onsets.
Conclusions
This concludes my series on how seed oils cause reductive stress. In part one we saw that mitochondria need proper oxygenation to burn cleanly. WIthout that, a flooded engine leads to rising levels of unburnt fuel including acetyl-CoA. In part two we looked at the superoxide/Glutathione Reductase/NNT pathway that ensures proper fuel oxygenation when we’re burning saturated fat but not when we burn seed oils. Part three detailed how rising acetyl-CoA levels activate lipogenic enzymes. Activated lipogenic enzymes represent a choice by the organism to store fat even if it doesn’t eat more calories. Part four detailed a strange behavior of seed oils – they drive so much reductive stress that lipogenic enzymes are rapidly turned on and they are rebuilt as saturated fat! Part five demonstrated how in the very short term, seed oils cause reductive stress in lean humans, causing them to behave metabolically like obese humans.
This last article showcases how oxidized PUFA act as activators of nuclear receptors which are known obesogens. Not only that, they are also oxidized by the cytochrome p450 enzymes that are upregulated by the nuclear receptors. This is a positive feedback loop ending in fat mice who ate soybean oil.
The whole series of events from ingestion of PUFA to obesity is a series of self-reinforcing control loops. When the animal chooses to fatten up by eating PUFA, it is the prerogative of the organism to make sure this happens.
ONE LAST NOTE: Some in this space have suggested that the effects of seed oils I am attributing to a change in mitochondrial dynamics are actually brought about by a rise in damaging oxylipins like 4-HNE. I believe that 4-HNE is another damaging element of PUFA consumption, but I didn’t feel the need to bring them up here since the dynamics I’m talking about can be largely explained without them IMO and Tucker Goodrich covers this topic well over at his blog Yelling Stop.
I’ve spent quite a bit of time examining in detail the effects of PUFA on mitochondrial dynamics. Next up, we’re going to look at how to break out of this self-reinforcing cycle! Probably after a brief solstice break, right after the 4th of July. Stay tuned!
- 1.Geiger SD, Yao P, Vaughn MG, Qian Z. PFAS exposure and overweight/obesity among children in a nationally representative sample. Chemosphere. Published online April 2021:128852. doi:10.1016/j.chemosphere.2020.128852
- 2.Sui Y, Ai N, Park SH, et al. Bisphenol A and Its Analogues Activate Human Pregnane X Receptor. Environmental Health Perspectives. Published online March 2012:399-405. doi:10.1289/ehp.1104426
- 3.RUBENSTRUNK A, HANF R, HUM D, FRUCHART J, STAELS B. Safety issues and prospects for future generations of PPAR modulators. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids. Published online August 2007:1065-1081. doi:10.1016/j.bbalip.2007.02.003
- 4.Baker AH, Watt J, Huang CK, Gerstenfeld LC, Schlezinger JJ. Tributyltin Engages Multiple Nuclear Receptor Pathways and Suppresses Osteogenesis in Bone Marrow Multipotent Stromal Cells. Chem Res Toxicol. Published online May 13, 2015:1156-1166. doi:10.1021/tx500433r
- 5.Girer NG, Tomlinson CR, Elferink CJ. The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with Ahr Ligands. IJMS. Published online December 23, 2020:49. doi:10.3390/ijms22010049
- 6.Xu C, Li CYT, Kong ANT. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. Published online March 2005:249-268. doi:10.1007/bf02977789
- 7.Hankinson O. The role of AHR-inducible cytochrome P450s in metabolism of polyunsaturated fatty acids. Drug Metabolism Reviews. Published online June 30, 2016:342-350. doi:10.1080/03602532.2016.1197240
- 8.HAN J, SUN L, XU Y, LIANG H, CHENG Y. Activation of PPARγ by 12/15-lipoxygenase during cerebral ischemia-reperfusion injury. International Journal of Molecular Medicine. Published online November 12, 2014:195-201. doi:10.3892/ijmm.2014.1998
- 9.Barquissau V, Ghandour RA, Ailhaud G, et al. Control of adipogenesis by oxylipins, GPCRs and PPARs. Biochimie. Published online May 2017:3-11. doi:10.1016/j.biochi.2016.12.012
- 10.Bock KW. Aryl hydrocarbon receptor (AHR) functions in NAD+ metabolism, myelopoiesis and obesity. Biochemical Pharmacology. Published online May 2019:128-132. doi:10.1016/j.bcp.2019.02.021
- 11.Song EK, Lee YR, Kim YR, et al. NAADP Mediates Insulin-Stimulated Glucose Uptake and Insulin Sensitization by PPARγ in Adipocytes. Cell Reports. Published online December 2012:1607-1619. doi:10.1016/j.celrep.2012.10.018
- 12.Beceiro S, Pap A, Czimmerer Z, et al. Liver X Receptor Nuclear Receptors Are Transcriptional Regulators of Dendritic Cell Chemotaxis. Mol Cell Biol. Published online May 15, 2018. doi:10.1128/mcb.00534-17
- 13.Zhou T, Cong S, Sun S, et al. Identification of endocrine disrupting chemicals activating SXR-mediated transactivation of CYP3A and CYP7A1. Molecular and Cellular Endocrinology. Published online January 2013:36-43. doi:10.1016/j.mce.2012.09.001
- 14.Chiaro ChristopherR, Patel RD, Perdew GaryH. 12(R)-Hydroxy-5(Z),8(Z),10(E),14(Z)-eicosatetraenoic Acid [12(R)-HETE], an Arachidonic Acid Derivative, Is an Activator of the Aryl Hydrocarbon Receptor. Mol Pharmacol. Published online September 8, 2008:1649-1656. doi:10.1124/mol.108.049379
- 15.Risérus U, Tan GD, Fielding BA, et al. Rosiglitazone Increases Indexes of Stearoyl-CoA Desaturase Activity in Humans. Diabetes. Published online May 1, 2005:1379-1384. doi:10.2337/diabetes.54.5.1379
- 16.Rojas IY, Moyer BJ, Ringelberg CS, Tomlinson CR. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of CYP1B1, PPARα, SCD1, and osteopontin. Int J Obes. Published online January 7, 2020:948-963. doi:10.1038/s41366-019-0512-z
- 17.Diani-Moore S, Ram P, Li X, et al. Identification of the Aryl Hydrocarbon Receptor Target Gene TiPARP as a Mediator of Suppression of Hepatic Gluconeogenesis by 2,3,7,8-Tetrachlorodibenzo-p-dioxin and of Nicotinamide as a Corrective Agent for This Effect. Journal of Biological Chemistry. Published online December 2010:38801-38810. doi:10.1074/jbc.m110.131573
Thank you for your posts Brad!
So far I’ve been taking ALA and berberine with great weight-loss success.
Anyway, I was listening to one of your interviews, you mentioned a study involving people training to run a marathon and their caloric expenditure. And how at the end of the study they were only burning something like 200 more calories per day from tons of this marathon training. I’ve done a lot of googling but I can’t seem to find the study, do you mind sharing the link by chance?
I got that from Herman Pontzer’s book “Burn”. I talk about it here:
https://fireinabottle.net/pontzers-burn-and-metabolic-rate-mechanisms/
Great work Brad, but I have little problem with your Level 1 explanation. Show me a paper, where LA cause less superoxide production than other fats. This is simply not true and I offer other explanation. The “overeating” of the cell is caused by overexpressing of GPx caused by free LA. All FFA increase GPx but LA far the most. This means that LA produce more O2- but SOD is slowed down and H2O2 is rapidly cleaned. This is special status of low H2O2 and high O2-. This has consequences and two phazes:
1. Very low insulin resistance while GPx is working and GSH is not depleted. There is lots of NAD+ for beta oxidation. Low H2O2, bad reaction to hypoxia (necrosis, inflamation).
2. High insulin resistance when GSH is depleted and GPx is not functional. There is lack of NAD+. High H2O2, pseudohyposia with restricted beta oxidation.
In both phazes there is plenty of O2- which slows down aconitase and all TCA cycle. This elevates acetylCoA, NAD+ is plenty in first phase and there is wild beta oxidation to more elevate acetylCoA, NAD+ is depleted after Complex 1 is blocked by GSH depletion. It is direct and protective mechanism.
At the point of the mitochondrial bottleneck, linoleic acid absolutely drives less ROS than saturated fat:
https://www.cellphysiolbiochem.com/Articles/000282/
That doeesn’t mean that ultimately a high LA doesn’t lead to a cell in oxidative stress. Very different things.
Brad
so i had googled once, on how animals get out of hibernation, and they do it by shaking, tremors, you know
anyway, regarding that, what do you think about silly devices like this one? https://youtu.be/xtUTku_LBSY
thanks, Brad
I doubt that has the same effects as shivering: driven by your internal muscle spasms.
Question for you, are you familiar with Morley Robbins and his work on iron & copper? He talks a lot about the importance of copper to mitochondrial function and about how it handles the oxygen. I was wondering what you have to say about that and if you agree with him and think the iron/copper dynamic is an important one. His take on health is that most people have too much iron stored in their tissues which causes oxidative stress and is unusable because we lack bioavailable copper and for both reasons (too much iron and too little copper) our mitochondria don’t work well. He doesn’t advocate seed oils but neither does he focus on them.